PRAVILNIKO METODAMA UZIMANJA UZORAKA I METODAMA FIZIČKIH, HEMIJSKIH I MIKROBIOLOŠKIH ANALIZA STOČNE HRANE("Sl. list SFRJ", br. 15/87) |
Kontrola kvaliteta stočne hrane, u smislu ovog pravilnika, vrši se na uzorcima za ispitivanje uzetim po metodama koje su propisane ovim pravilnikom.
Metode kojima se vrši kontrola kvaliteta stočne hrane su:
1) metoda uzimanja uzoraka;
2) metoda fizičkih i hemijskih analiza;
3) mikrobiološka metoda.
Metodama uzimanja uzoraka stočne hrane utvrđuju se postupci i način uzimanja uzoraka čiji se kvalitet kontroliše.
Metodama fizičkih i hemijskih analiza utvrđuju se uslovi i postupci za pripremanje laboratorijskog uzorka i fizičke i hemijske analize stočne hrane, radi provere fizičkih svojstava i hemijskog sastava tih proizvoda.
Mikrobiološkim metodama utvrđuje se sadržaj antibiotika u stočnoj hrani.
U izveštaju o fizičkoj i hemijskoj analizi moraju biti prikazani rezultati utvrđeni primenom metoda fizičkih i hemijskih analiza propisanih ovim pravilnikom.
Rezultati se daju kao aritimetička sredina najmanje dva određivanja koja je, istovremeno ili neposredno jedno za drugim, obavio isti analitičar u istoj laboratoriji.
METODE UZIMANJA UZORAKA STOČNE HRANE
Uzimanje uzoraka stočne hrane, shodno ovom pravilniku, mora da izvrši stručno lice.
Uzimanje uzoraka stočne hrane vrši se:
- u proizvodnji, na proizvodnoj partiji ili delu proizvodne partije;
- u prometu, na jedinicama pošiljke.
Uzimanje uzoraka stočne hrane u proizvodnji i prometu mora se izvršiti tako da svaka jedinica proizvoda ima istu mogućnost da bude izabrana kao uzeti uzorak.
Način uzimanja uzorka mora biti isti u proizvodnji i u prometu.
Uzorak mora predstavljati prosečan sastav celokupne količine proizvoda od koje se uzima uzorak.
Pod isporukom (pošiljkom) stočne hrane podrazumeva se određena količina stočne hrane pripremljena za stavljanje u promet.
Pod partijom stočne hrane podrazumeva se količina istog proizvoda određene isporuke koja ima isti kvalitet.
Pod pojedinačnim (osnovnim) uzorkom podrazumeva se manja količina proizvoda uzeta sa jednog mesta partije.
Serija pojedinačnih uzoraka mora biti uzeta sa različitih mesta iz proizvodne partije.
Pod zajedničkim (zbirnim) uzorkom podrazumeva se skup sjedinjenih i brižljivo izmešanih pojedinačnih uzoraka jedne partije.
Pod prosečnim uzorkom podrazumeva se uzorak koji se dobija redukcijom zajedničkog (zbirnog) uzorka i služi za laboratorijsko ispitivanje.
Broj jedinica uzetog uzorka zavisi od vrste proizvoda, mase, odnosno zapremine proizvoda i količine pošiljke.
Pojedinačni uzorci zrnaste, brašnaste i slične stočne hrane koja se isporučuje ili skladišti u rasutom stanju uzimaju se:
1) iz vagona, kamiona i sl, sa najmanje osam mesta ravnomerno raspoređenih po površini, i to: sa vrha, iz sredine i sa dna;
2) iz šlepova i brodova za vreme utovara, odnosno istovara, na način iz stava 2. ovog člana;
3) iz skladišta, na odstojanju od najviše dva metra između pojedinih mesta sondiranja, s tim da se iz gornjeg i donjeg sloja uzima isti broj uzoraka kad visina uskladištene mase nije veća od 0,75 m, a iz gornjeg, srednjeg i donjeg sloja - kad je visina uskladištene mase veća od 0,75 m.
Pri pneumatskom utovaru ili istovaru rasutog materijala uzorci se uzimaju u istim vremenskim razmacima, na najpogodnijim radnim mestima, izdvajanjem pojedinačnih uzoraka u približno istim količinama.
Pojedinačni uzorci od briketirane stočne hrane i uljanih pogača uzimaju se kao i za zrnasta hraniva, s tom razlikom što se od briketa i pogača uzima po jedan deo, a sitnije presovani oblici (kockice, palete, granule) tretiraju se kao zrnasta i slična hraniva.
Pojedinačni uzorci stočne hrane koja je upakovana u vrećama bez obzira na veličinu vreće, uzimaju se sa vrha, iz sredine i sa dna vreće.
Broj uzetih pojedinačnih uzoraka zavisi od veličine isporuke.
Ako isporuka u rasutom stanju, odnosno jedinica isporuke ili partija proizvoda ne iznosi više od 10 tona, uzima se najmanje 20 pojedinačnih uzoraka, čija ukupna masa mora da iznosi najmanje 3 do 5 kg.
Od svake sledeće ili započete količine od 10 tona stočne hrane uzima se još po 2 kg u pojedinačnim uzorcima.
Kad je stočna hrana pakovana u vrećama, pojedinačni uzorci uzimaju se, i to:
- od partije do 20 tona - iz svake desete vreće, uzete po slobodnom izboru, a iznad te količine - iz svake dvadesete vreće;
- za količine stočne hrane manje od 10 tona - veći pojedinačni uzorci, tako da njihova masa iznosi 3 do 5 kg;
- za količine stočne hrane do 500 kg - pojedinačni uzorci iz svake vreće.
Svi pojedinačni uzorci stavljaju se u podesan čist i suv sud, a briketirana i presovana hraniva prethodno se dobro usitne i izmešaju u zajednički uzorak.
Kad je u pitanju pneumatski transport, jedan zajednički uzorak formira se iz količine do 100 tona.
Kad je u pitanju transport brodom, odnosno drugi vidovi i načini isporuke, jedan zajednički uzorak se formira iz količine do 30 tona.
Redukcijom zajedničkog uzorka dobijaju se prosečni uzorci za analizu.
Zajednički uzorak postavi se na ravnu i čistu podlogu i uobliči u piramidu koja se pritiskom odozgo zaravni. Dobijena zarubljena piramida dijagonalno se podeli na bočne strane i izdvoje se naizmenični segmenti. Sa izdvojenom količinom postupa se na isti način sve dok se od zajedničkog uzorka ne dobije masa od 3 do 5 kg.
Na taj način dobijena masa uzorka deli se na tri prosečna uzorka za analizu čija masa mora da iznosi najmanje 1 kg.
Prosečan uzorak za laboratorijsko ispitivanje mora se pripremiti u najmanje dva primerka, u količini koja je potrebna za fizičke, hemijske i mikrobiološke analize, s tim da sve jedinice laboratorijskog uzorka moraju biti identične po sastavu, a približno jednake po masi. Od uzetih uzoraka, jednu jedinicu uzorka dostavlja na analizu stručno lice koje uzima uzorak, dok druga jedinica uzorka služi za superanalizu.
Na zahtev predstavnika organizacije udruženog rada mora se uzeti i treći primerak, koji se dostavlja na raspolaganje tom licu.
Uzorci se pakuju u čiste i suve posude od stakla, pocinkovanog lima, aluminijuma, plastičnog ili drugog pogodnog materijala koji ne oksidiše i ne utiče na promenu sastava i kvaliteta uzetih uzoraka i koje se mogu hermetički zatvarati.
Posude sa prosečnim uzorkom osiguravaju se kanapom, kojim se fiksira i poseban karton sa deklaracijom (privezak, etiketa).
Krajevi kanapa učvršćuju se pečatnim voskom da bi se originalni uzorak obezbedio, odnosno da bi se onemogućilo otvaranje uzorka bez povrede i oštećenja pečata i pakovanja.
Kad se završi uzimanje uzoraka sastavlja se zapisnik, koji potpisuju lica koja su uzimala uzorke.
Zapisnik mora da sadrži sva zapažanja o stanju u kome se proizvod nalazi u trenutku uzimanja uzorka, pri čemu se naznačuju vidljivi tragovi oštećenja nastalih u skladištu, odnosno silosu ili oštećenja nastala tokom manipulacije u brodu ili nekom drugom prevoznom sredstvu.
U zapisniku se navodi i način uzimanja uzorka i sve okolnosti koje mogu uticati na njihovo uzimanje.
Uz svaki uzorak za ispitivanje mora se priložiti zapisnik o uzimanju uzoraka i deklaracija proizvođača. U zapisnik se upisuju sledeći podaci:
1) naziv hraniva;
2) prevozno sredstvo i njegova oznaka (broj);
3) naziv mesta iz koga je isporučeno;
4) naziv mesta u koje je isporuka upućena;
5) datum prispeća isporuke;
6) količina stočne hrane;
7) broj vreće ili oznaka "rasuto";
8) oznaka za identifikaciju ili broj partije;
9) firma, odnosno naziv i sedište proizvođača, odnosno isporučioca;
10) datum završetka utovara, odnosno istovara;
11) bliža oznaka mesta i datum uzimanja uzorka;
12) potpis lica koje je uzelo uzorak.
Podaci u deklaraciji moraju biti neizbrisivi.
Uzeti prosečni uzorci šalju se na ispitivanje odmah ili najkasnije 48 h posle uzimanja uzoraka.
O uzetom uzorku mora se sačiniti zapisnik.
Prosečni uzorci čuvaju se najmanje 60 dana.
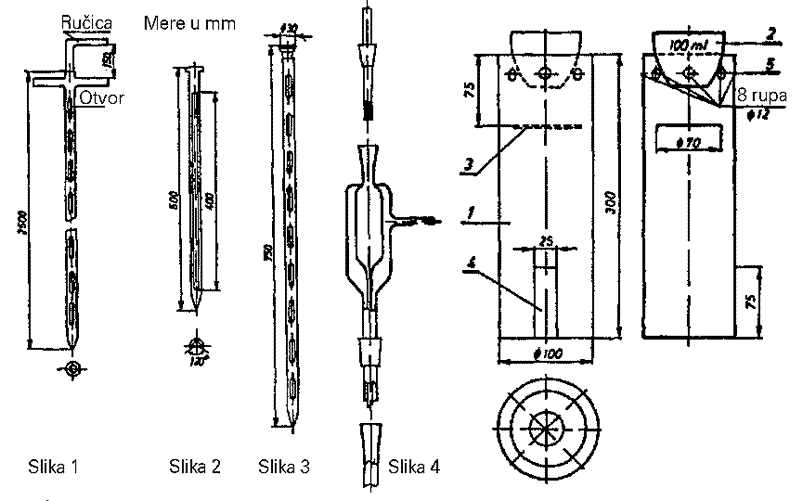
Za uzimanje uzorka koriste se sonde cilindričnog oblika raznih veličina koje se sastoje od jedne cevi i sonde sa dve cevi (sonde sa odeljcima).
Sonda sa odeljcima (sl. 1 u prilogu) upotrebljava se kad se uzorci uzimaju iz hraniva u rasutom stanju, u mirovanju. Ona se sastoji od dve koncentrične cevi koje ulaze jedna u drugu.
Spoljašnja cev ima na gornjem kraju dvokraku ručicu, otvor duž cele cevi i otvore koji odgovaraju malim odeljcima u unutrašnjoj cevi. Unutrašnja cev ima jednakokraku ručicu i nekoliko otvora po celoj dužini. Odeljci u cevi onemogućavaju da se proizvod u njima meša.
Sonde za uzimanje uzoraka iz vreća su cilindrične, od poliranog metala. Na vrhu imaju zašiljeni deo za probijanje, čija dužina iznosi najmanje 25 cm. Sonde mogu biti načinjene od jedne cevi (sl. 2 u prilogu) ili od dve cevi koje ulaze jedna u drugu (sl. 3 u prilogu) kao sonda sa odeljcima. Veličina proreza na sondama iznosi najmanje trećinu površine cilindra.
METODE FIZIČKIH, HEMIJSKIH I MIKROBIOLOŠKIH ANALIZA STOČNE HRANE
Metode fizičkih, hemijskih i mikrobioloških analiza kojima se ispituje kvalitet stočne hrane su:
1) pripremanje laboratorijskog uzorka;
2) određivanje mirisa;
3) određivanje količine primesa;
4) dokazivanje zaraženosti štetočinama;
5) određivanje zapreminske mase;
6) određivanje sadržaja vlage;
7) određivanje sirovih proteina;
8) određivanje amonijačnog azota;
9) određivanje uree - spektrofotometrijska metoda;
10) određivanje kazeina;
11) određivanje laktoalbumina;
12) određivanje sirovih masti;
13) određivanje slobodnih masnih kiselina;
14) određivanje kiselinskog stepena;
15) određivanje pH vrednosti;
16) određivanje sirove celuloze;
17) određivanje skroba;
18) određivanje sirovog pepela;
19) određivanje pepela nerastvorljivog u hlorovodoničnoj kiselini;
20) određivanje bezazotnih ekstraktivnih materija;
21) određivanje hlorida;
22) određivanje natrijum-hlorida;
23) određivanje organoleptičkih svojstava jodirane soli za stočnu hranu;
24) određivanje vode u jodiranoj soli (higroskopnoj i kristalnoj);
25) određivanje materija nerastvorljivih i rastvorljivih u vodi u jodiranoj soli;
26) određivanje natrijuma i kalijuma;
27) određivanje kalcijuma - kompleksometrijska metoda;
28) određivanje magnezijuma;
29) određivanje ukupnog fosfora - spektrofotometrijska metoda;
30) određivanje sumpora;
31) određivanje hlora;
32) određivanje joda;
33) određivanje mikroelemenata - spektrofotometrijska metoda;
34) određivanje mangana - spektrofotometrijska metoda;
35) određivanje kobalta - spektrofotometrijska metoda;
36) određivanje gvožđa - spektrofotometrijska metoda;
37) određivanje cinka - spektrofotometrijska metoda;
38) određivanje bakra - spektrofotometrijska metoda;
39) određivanje karotina (brzi postupak);
40) određivanje karotina i ksantofila u svežim i suvim uzorcima biljnog porekla;
41) određivanje vitamina A;
42) određivanje vitamina E;
43) određivanje mikotoksina;
44) određivanje aktivnosti antibiotika mikrobiološkim metodama na Petrijevim šoljama (metoda difuzije);
45) određivanje oksitetraciklin-hidrohlorida (OTC HCl);
46) određivanje hlortetraciklin-hidrohlorida (CHTC HCl);
47) određivanje oleandomicina;
48) određivanje benzatin penicilina G;
49) određivanje cink-bacitracina;
50) određivanje tilozina;
51) određivanje neomicina;
52) određivanje virginiamicina.
Metode fizičkih, hemijskih i mikrobioloških analiza za kontrolu kvaliteta stočne hrane odštampane su uz ovaj pravilnik i čine njegov sastavni deo.
Posle ispitivanja proizvoda sačinjava se zapisnik - izveštaj o ispitivanju u kome se navodi primenjena metoda, dobijeni rezultat i uslovi ispitivanja, kao i okolnosti koje su mogle uticati na rezultat.
Zapisnik o ispitivanju mora sadržati sve podatke za potpunu identifikaciju proizvoda.
Ovaj pravilnik stupa na snagu po isteku tri meseca od dana objavljivanja u "Službenom listu SFRJ".
METODE FIZIČKIH, HEMIJSKIH I MIKROBIOLOŠKIH ANALIZA STOČNE HRANE
1. Pripremanje laboratorijskog uzorka
Laboratorijski uzorak, odnosno uzorak za analizu dobija se iz prosečnog uzorka redukcijom i isitnjavanjem, eventualnim sušenjem i odmašćivanjem, zavisno od vrste stočne hrane.
Odredbe iz stava 1. odnose se na kabasta hraniva, koncentrate, žitarice, zrna leguminoza i slične proizvode.
Reagensi
Za pripremanje laboratorijskog uzorka koriste se sledeći reagensi i destilovana voda:
1) dietil-etar bezvodni, bez peroksida (r20 = 0,720 g/ml, tačke ključanja 34 do 35°C);
2) n-heksan;
3) diatomejska zemlja.
Pribor
Za pripremanje laboratorijskog uzorka koristi se sledeći pribor:
1) naprava za deobu uzorka (razdeljivač);
2) sekač;
3) makaze;
4) prekrupač;
5) strugač;
6) mehanička mešalica;
7) avan ili kuglični mlin;
8) mlin sa sitom sa otvorima veličine 0,5 mm;
9) analitička vaga;
10) sušnica sa ventilatorom i regulacijom temperature;
11) plitka posuda od aluminijuma.
Određivanje
Deoba prosečnog uzorka
Pomoću naprave za deobu prosečan uzorak se podeli na dva dela. Kabasta i sveža hrana koje se ne mogu deliti napravom dele se ručno, i to tako što se ceo prosečan uzorak stavi na ravnu i suvu podlogu, a zatim rasporedi u obliku kruga ili kupe. Tako pripremljen uzorak podeli se po dijagonali na četiri dela, pa se dva suprotna odeljka sjedine u prvi deo uzorka (A), a ostala dva - u drugi deo uzorka (B).
Ako je uzorak veliki, a to dopušta njegova homogenost, uzorak (B) može se pre isitnjavanja još jedanput podeliti (pri čemu se drugi deo odbaci), tako da se svodi na 100 do 500 g mase.
Deo uzorka (A) koristi se za analize koje se izvode na nepromenljivom materijalu (analiza prosejavanjem, analiza na organske kiseline, na pH vrednost i druge analize), a čuva se i kao rezerva do kraja analize.
Deo uzorka (B), direktno ili po prethodnom sušenju ili odmašćivanju, isitni se i upotrebi za hemijske analize.
Pri deobi se vodi računa o tome da ne dođe do izmene uzorka i promene sadržaja vlage.
Prosečni uzorci kabaste hrane (seno, silaža, sveža trava, klipovi kukuruza i sl) pre deobe se iseku na kraće komade kako bi se dobili delovi jednaki po količini i identični u pogledu sastava.
Pripremanje uzorka sa visokim sadržajem vlage
Stočna hrana sa visokim sadržajem vlage teško se melje, pa je neophodno uzorak za analizu posebno pripremiti sušenjem. To sušenje se mora izvoditi u dve faze.
U prvoj fazi, pripremljeni uzorak se unosi u sušnicu u kojoj je temperatura od 60 do 70°C i pojačana cirkulacija vazduha (ugrađen ventilator ili odškrinuta vrata). Takvo sušenje traje dok se sadržaj vlage ne svede na 7 do 10%. Posle toga uzorci se vade iz sušnice i u otvorenim posudama u kojima su sušeni ostavljaju najmanje 1 h na sobnoj temperaturi. Za to vreme postiže se ravnotežna vlažnost. Sledeći postupak je mlevenje tako osušenih uzoraka i prenošenje u sudove koji se dobro zatvaraju, čime se onemogućava dalje povećanje vlage.
Na osnovu gubitka mase u toku prve faze sušenja, odnosno postizanja ravnotežne vlažnosti, izračunava se sadržaj vlage ili vazduhom sušene materije (VSM).
U drugoj fazi uzorak se suši do postizanja konstantne mase, u sušnicama na temperaturi 105 °C, prema metodi za određivanje sadržaja vlage u stočnoj hrani.
Količina suve materije u originalnom uzorku u procentu mase izračunava se po formuli:
SM = |
VSM · SM´ |
|
100 |
gde je:
SM - |
suva materija u originalnom uzorku, u procentima; |
VSM - |
vazduhom sušena materija, u procentu mase, određena prilikom pripremanja uzorka; |
SM´ - |
suva materija određena na 105°C od vazduhom sušene materije, u procentu mase. |
Analitički podaci, određeni kasnije u osušenom i samlevenom materijalu, preračunavaju se na originalni uzorak množenjem konverzionim faktorom F = SM/SM´.
Napomena. |
Ovom metodom ponekad treba osušiti i uzorke sena. |
Isitnjavanje uzorka
Uzorak sa niskim sadržajem vlage (deo B) ceo se isitnjava posle deobe prosečnog uzorka.
Uzorak sa visokim sadržajem vlage, posle sušenja i merenja, sjedini se i samelje.
Uzorci se isitne tako da sve čestice prođu kroz sito veličine otvora 0,5 mm. Za specijalne analize, gde su mase koje se mere manje od 1 g, reprezentativan deo tako isitnjenog uzorka i dalje se sitni, tako da sve čestice uzorka prođu kroz sito veličine otvora 0,1 mm.
Isitnjavanje se vrši u avanu ili laboratorijskom mlinu što je moguće brže, uz što manje izlaganja uzorka vazduhu, koje bi moglo da prouzrokuje nedozvoljen gubitak ili povećanja vlage.
Napomena. |
Mogućnost gubitka isparljivih materija veća je pri prosejavanju uzorka isitnjenog na mlinu nego ručno, što se pri analizi mora uzeti u obzir. |
Isitnjen uzorak se dobro promeša i prenese u sud koji ne korodira i koji se može dobro zatvoriti. Čuva se na suvom, hladnom i tamnom mestu, na sobnoj temperaturi, i služi za hemijsku analizu.
Uzorci sa visokim sadržajem masti i drugi uzorci
Ako se zbog visokog sadržaja masti uzorak ne može dobro samleti, on se grubo samelje u pogodnom mlinu, izdrobi ili istruže. Uzorak se izmeri po postupku pripremanja uzorka sa visokim sadržajem vlage, s tačnošću 0,1 g, i najpre osuši, a zatim se ekstrahuje sa dietil-etrom ili n-heksanom. Rastvor, sa rastvorenom mašću, sakupi se u izmerenu tikvicu, a rastvarač se predestiliše i, posle sušenje 1 h na temperaturi 103°C, tačno se utvrdi količina masti.
Uzorak se ostavi nekoliko sati na vazduhu da bi rastvarač ispario i da bi primio ravnotežnu količinu vlage, zatim se izmeri i isitni, a posle toga upotrebi za analizu.
Rezultati se moraju preračunati na početni materijal, množenjem konverzionim faktorom f = m1/m0,
gde je:
m0 - |
masa originalnog uzorka za sušenje i odmašćivanje, u gramima; |
m1 - |
masa uzorka odmašćenog i sušenog na sobnoj temperaturi, u gramima. |
Pri određivanju masti u uzorku uzima se u obzir i količina masti određena pri pripremanju uzorka.
Uzorke koji se teško suše zbog sloja masti koji prekriva vlažnu površinu pre sušenja treba pomešati sa određenom količinom diatomejske zemlje ili kvarcnog peska. Količina diatomejske zemlje ili kvarcnog peska zavisi od sadržaja vode i masti.
Napomena. |
Diatomejska zemlja i kvarcni pesak ponašaju se kao inertni materijal i određuju se kao deo pepela. |
Rezultati ove analize moraju se korigovati prema količini dodate diatomejske zemlje ili kvarcnog peska.
Analiza melase vrši se na svežim uzorcima.
Šećerna repa, krompir i slični proizvodi izrežu se na tanke delove i osuše. Ako postoje odgovarajući uređaji, mogu se i homogenizovati do kaše, koja se direktno uzima za pojedinačna određivanja. Ako je količina suve materije mala i homogenost slaba, uzorak se mora povećati.
Prilikom sušenja uzorka silaže prema postupku pripremanja uzorka sa visokim sadržajem vlage, osim vode, gube se i druge isparljive materije, pa se pri određivanju suve materije mora vršiti korekcija prema obrascu:
SMk = SM + (OC + PR + MA)
gde je:
SMk - |
korigovana suva materija, u procentima; |
SM - |
suva materija, izračunata prema postupku pripremanja uzorka sa visokim sadržajem vlage, u procentima; |
OC + PR + MA - |
zbir sirćetne, propionske i buterne kiseline, određen u originalnom uzorku, u procentima. |
Napomena. |
Da bi se prilikom sušenja uzorka sprečilo isparavanje isparljivih masnih kiselina, što utiče na rezultat određivanja suve materije, uzorak se posle merenja, pre sušenja, navlaži tankim mlazom 10%-nog rastvora natrijum-hidroksida. Količina natrijum-hidroksida mora biti poznata i uzima se u obzir pri proračunavanju suve materije. |
Primena
Metoda određivanja mirisa primenjuje se za žita, leguminoze i mlinske proizvode od žita i leguminoza namenjenih za stočnu hranu.
Određivanje
Oko 20 g uzorka žita ili leguminoza prekrupi se na potpuno čistom laboratorijskom mlinu, koji mora biti bez mirisa. Dobijena prekrupa ili ista količina mlinskih proizvoda žita ili leguminoza prenese se u čašu, prelije toplom vodom čija temperatura iznosi 60°C, zatim se voda izlije, a ostatak ispituje organoleptički. Ako najmanje tri od pet degustatora utvrde neki od stranih mirisa navedenih u propisu o kvalitetu stočne hrane, smatraće se da su ti mirisi utvrđeni.
3. Određivanja količine primesa
Izdvajanje metalnih primesa
Od prosečnog uzorka izmeri se 100 g, sa tačnošću 0,01 g, razastre se na plitku ravnu površinu (najbolje staklenu) u ujednačenom tankom sloju, visine najviše do 0,5 cm, pa se laganim kretanjem magneta uzduž i popreko, neposredno nad površinom sloja, izdvoji metal iz uzorka, uz nastojanje da cela površina bude zahvaćena magnetskim poljem. Povremeno se sa magneta otklone (oduvaju) nemetalni delovi (brašno, mekinje i sl), a delovi metala skinu na papir. Prevlačenje magneta preko uzorka ponavlja se tri puta. Pre svakog prevlačenja ispitivani uzorak se izmeša i poravna u tankom sloju. Sve metalne čestice skinute sa magneta skupe se na sahatno staklo, izmere na analitičkoj vagi i njihova količina izrazi u miligramima na 1 kg proizvoda.
Magnet koji se koristi za ovo određivanje treba da se koristi samo u analitičke svrhe, a kad se ne upotrebljava, površine oba pola magneta treba da budu pokrivene gvozdenom pločicom debljine 10 mm. Magnetna sila ovog magneta treba da iznosi najmanje 1,2 N.
Posle izdvajanja metalnih delova, uzorak se ispituje na druge primese.
Određivanje ostalih primesa
Uzorak se razastre na ploču, pa se sve primese odvoje po vrstama, čiji je sadržaj propisan Pravilnikom o kvalitetu stočne hrane. Svaka primesa se posebno izmeri i sadržaj izrazi u procentima. Takođe se odredi zbir svih primesa, izražen u procentima.
Određivanje mineralnih primesa
U čist i suv levak za odvajanje stavi se 10 ml hloroforma i doda se 50 g uzorka izmerenog sa tačnošću 0,01 g, iz koga su prethodno izdvojene metalne primese i staklo. Pošto se protrese nekoliko puta tečnost u levku za odvajanje ostavi se da miruje 10 min da bi se mineralne nečistoće izdvojile i pale na dno. Tada se nečistoće ispuste u porculansku posudu, prethodno sušenu 15 min na temperaturi 130°C i, posle hlađenja izmere sa tačnošću 0,01 g. Nečistoće se ispuštaju otvaranjem slavine na levku nekoliko puta, pri čemu se slavina okreće za 360°C. Ako su delići nečistoće veći od otvora slavine, izvade se pincetom kroz gornji otvor levka i stave u posudu sa ispuštenim nečistoćama; doda se malo hloroforma, ispari na vodenom kupatilu, suši i žari 1 h na temperaturi 550°C, hladi u eksikatoru i izmeri sa tačnošću 0,01 g.
Sadržaj mineralnih nečistoća (x), osim stakla i metala, izračunava se po sledećoj formuli:
x = (a - b) · 2
gde je:
a - masa posude u sa nečistoćama, u gramima;
b - masa posude, u gramima.
Kao rezultat određivanja uzima se srednja aritmetička vrednost rezultata iz najmanje tri određivanja.
4. Dokazivanje zaraženosti štetočinama
Izmeri se 0,5 do 1 kg uzorka za ispitivanje, proseje se kroz sito sa okruglim otvorima prečnika 2 mm. Ostaci posle prosejavanja rasprostru se u tankom sloju na beli papir ili u široku posudu i pažljivo ocenjuju.
U tim ostacima utvrđuju se prisustvo i vrste krupnijih živih štetočina i njihovih larvi (žižak, brašnari, moljci i dr), a u propadu gušćeg sita - sitnije štetočine (larve, jaja, delovi i izmet krpelja, brašnara i dr). To se utvrđuje tako što se na ravnoj podlozi, pod pritiskom staklene ploče ili papira, prosejano brašno ili mekinje poravnaju u tankom sloju, debljine 1 do 2 mm i površina pažljivo posmatra. Izdignuća i brazda na površini brašna ukazuju na zaraženost krpeljima. Temperatura uzorka pri ispitivanju treba da iznosi 15 do 18°C.
5. Određivanje zapreminske mase
Princip i primena
Metoda se zasniva na određivanju zapisnika zapreminske mase žita izražene u kg/m3. Određivanje zapreminske mase primenjuje se pri ispitivanju žita i leguminoza za stočnu hranu.
Provera vage
Pre početka rada proveri se tačnost vage tako što se na jednu stranu obesi merni cilindar u kome se nalazi klip, a na drugu - tas za stavljanje tegova. Zatim se merni cilindar skine sa vage i iz njega se izvadi klip. Cilindar se stavlja na postolje, a zatim se kroz njegov prorez uvlači nož, na koji se stavi klip. Na merni cilindar učvrsti se cev za nasipanje. Na taj način vaga je pripremljena za rad.
Određivanje
Uzorak za ispitivanje rasprostre se po površini stola i podeli četvrtanjem. Zatim se iz svakog kvadrata lopaticom uzima jednaka količina uzorka i stavlja u cev za nasipanje, do urezane crte. Sa odstojanja od 4 cm od vrha cilindra sipa se žito iz cevi u cilindar takvom brzinom da se cilindar zapremine 0,250 l napuni za 8 sekundi. Mlaz žita mora padati u sredinu cilindra, a žito se ne sme izravnavati sa rubom cilindra. Uz pridržavanje mernog cilindra, nož se brzo izvuče, ali bez potresanja, pri čemu klip sa uzorkom naglo pada na dno mernog cilindra. Tada se nož ponovo uvuče u prorez, a cilindar se obesi na vagu i meri.
Izračunavanje
Zapreminska masa izražava se u kg/m3 i izračunava tako što se vrednost pročitana u tabeli pomnoži sa 10.
Princip i primena
Princip se sastoji u određivanju gubitaka mase sušenjem uzorka pod posebnim uslovima, zavisno od prirode uzorka, i izražava se u procentima mase.
Metoda se primenjuje za ispitivanje svih vrsta stočne hrane, izuzev za proizvode od mleka, mineralna hraniva i njihove smese.
Aparatura i pribor
Pored uobičajene laboratorijske opreme, koristi se sledeći pribor:
1) analitička vaga;
2) posude od nerđajućeg metala ili stakla, sa odgovarajućim poklopcem koji se dobro zatvara;
3) električna sušnica sa regulatorom temperature;
4) električna vakuum-sušnica;
5) eksikator sa efikasnim sredstvom za sušenje (kao CaCl2).
Pripremanje uzorka
Zavisno od vrste stočne hrane, uzorci za određivanje sadržaja vlage posebno se pripremaju kao što je propisano u metodi 1. ovog pravilnika - pripremanje laboratorijskog uzorka, i to:
- uzorci sa visokim sadržajem vlage (više od 17% m/m) i niskim sadržajem mulja prethodno se suše;
- uzorci sa visokim sadržajem masti i niskim sadržajem vlage prethodno se odmašćuju;
- uzorci sa visokim sadržajem masti i visokim sadržajem vlage prethodno se i suše i odmašćuju.
Određivanje
Posuda sa poklopcem najpre se suši 30 min na temperaturi 105°C i meri sa tačnošću 0,001 g. U posudu za sušenje odmeri se 5 g isitnjenog i homogenizovanog uzorka, sa tačnošću od 0,001 g, i ravnomerno rasprostre po dnu posude, a zatim se postupa kao što je određeno u daljem postupku.
Napomena. |
Kad su u pitanju hraniva u tečnom stanju ili u obliku paste i hraniva koja sadrže više masti i ulja, postupa se na sledeći način: u posudu se odmeri 10 g homogenizovanog uzorka, sa tačnošću od 0,001 g, i meša sa osušenim kvarcnim peskom sve dok pesak potpuno ne apsorbuje mast ili ulje. Posuda se ponovo izmeri, sa tačnošću od 0,001 g, zatim se stavi u peć i postupi kao što je određeno u daljem postupku. |
Posuda sa uzorkom stavi se u zagrejanu sušnicu regulisanu na temperaturu 105°C, a poklopac pored nje. Posuda se suši 4 h, odnosno do konstantne mase, na temperaturi 105°C, a zatim se poklopi i izvadi iz sušnice, stavi u eksikator, hladi 45 min i izmeri sa tačnošću 0,001 g. Pri tom se vodi računa o tome da za vreme merenja uzorak ponovo ne ovlaži.
Sušenje se ponovi pri temperaturi od 105°C u toku 2 h, i uzorak se 45 min hladi u eksikatoru. Ako je za vreme tog sušenja gubitak u masi sušenja veći od 0,2 %, znači da su nastale hemijske reakcije, odnosno gubitak suve materije, pa se u tom slučaju postupak sušenja obavlja u vakuum-sušnici.
Za hraniva koja sadrže više masti i ulja sušenje se ponavlja pri temperaturi 105°C u toku 30 min. Ako je gubitak u masi za vreme tog sušenja veći od 0,1 %, znači da su nastale hemijske reakcije, odnosno gubitak suve materije, pa se postupak obavlja u vakuum-sušnici.
Postupak sušenja u vakuum-sušnici
U prethodno osušenu posudu sa poklopcem i izmerenu sa tačnošću od 0,001 g odmeri se 5 g uzorka, sa tačnošću od 0,001 g, i ravnomerno rasprostre po dnu posude.
Posuda sa poklopcem pored nje stavi se u vakuum-sušnicu zagrejanu na temperaturu 80°C.
Pritisak se smanji na 13,33 kPa. Kad se taj pritisak postigne prekida se rad vakuum-pumpe. Uzorak se suši 4 h, na temperaturi 80°C, do konstantne mase.
Pritisak u sušnici pažljivo se podesi prema atmosferskom pritisku. Po otvaranju sušnice, posuda se brzo pokrije poklopcem, izvadi se iz peći i stavi u eksikator da se hladi 45 min i meri se sa tačnošću od 0,001 g. Posle toga, posuda sa uzorkom suši se još 30 min u vakuum-sušnici na temperaturi 80°C i meri. Razlika između dva merenja ne sme biti veća od 0,2 %.
Izračunavanje
Sadržaj vlage, izražen u procentima mase, izračunava se po formuli:
W = |
(ma - mb) · |
100 |
|
ma |
gde je:
ma - masa uzorka, u gramima;
mb - masa uzorka posle sušenja, u gramima.
Sadržaj vlage u uzorku sa visokim sadržajem vlage i niskim sadržajem ulja, u uzorku sa visokim sadržajem masti i niskim sadržajem vlage i uzorku sa visokim sadržajem masti i visokim sadržajem vlage izračunava se po sledećoj formuli:
|
(m0 - m3) - (m2 - |
W |
) |
|
W1 = |
100 |
· 100 |
||
m0 |
|
|||
gde je:
m0 - masa smanjenog uzorka pre mlevenja, u gramima;
m2 - masa ekstrahovanog i osušenog uzorka posle sušenja na sobnoj temperaturi, u gramima;
m3 - masa ekstrahovane masti iz smanjenog uzorka, u gramima;
10 m3 - masa alikvotnog dela ekstrahovane masti iz smanjenog uzorka, u gramima;
W - sadržaj vlage izražen u procentima mase ispitivanog uzorka.
Napomena. |
Za uzorke sa visokim sadržajem masti i niskim sadržajem vlage i za uzorke sa visokim sadržajem masti i visokim sadržajem vlage oznaka m3 mora se zameniti oznakom 10 m3. |
Prilikom sušenja uzoraka silaže prema postupku pripremanja uzoraka sa visokim sadržajem vlage, osim vode gube se i druge isparljive materije, pa se pri određivanju suve materije mora vršiti i korekcija prema obrascu:
SMk = SM - (OC + PR + MA)
gde je:
SMk - korigovana suva materija, u procentima;
SM - suva materija u procentima, izračunata prema postupku pripremanja uzorka sa visokim sadržajem vlage;
OC + PR + MA - zbir sirćetne, propionske i buterne kiseline, određen u originalnom uzorku, u procentima.
Osim računskim putem, pravi sadržaj suve materije, odnosno vlage može se odrediti na taj način što se uzo-rak posle merenja, a pre sušenja nakvasi određenom količinom 10 %-nog natrijum-hidroksida. Dodata količina natrijum-hidroksida mora se uzeti u obzir pri preračunavanju.
Kao rezultat analize uzima se srednja aritmetička vrednost dva uporedna ispitivanja ako su ispunjeni zahtevi u pogledu ponovljivosti.
Ponovljivost
Razlika između rezultata dva paralelna određivanja, koja je istovremeno ili jedno za drugim izveo isti anali-tičar, ne sme biti veća od 0,2 % (m/m).
7. Određivanje sirovih proteina
Princip i primena
Metoda se sastoji u razlaganju organske materije sumpornom kiselinom u prisustvu katalizatora. Razloženom uzorku dodaju se alkalije u višku, a oslobođeni amonijak se destiliše i titrira. Dobijeni sadržaj azota preračuna se faktorom 6,25 na sadržaj sirovih proteina.
Primenom ove metode ne dobija se razlika između proteinskog i neproteinskog azota. Ako je potrebno odre-đivanje sadržaja neproteinskog azota, primenjuje se druga metoda.
Reagensi i pomoćna sredstva
Za određivanje sirovih proteina, potrebni su sledeći reagensi, čistoće pro analysi, i destilovana voda:
1) kalijum sulfat, K2SO4;
2) sumporna kiselina, koncentrovana, r = 1,84 g/ml;
3) katalizatori:
- živa ili živin II-oksid (HgO) ili bakar II-oksid (CUO) ili bakar II-sulfat (CuSO4 · 5H2O);
4) parafinski vosak;
5) saharoza;
6) standardni rastvori:
- acetanilid (tačka topljenja 114°C) - sadrži azota 10,37 % m/m;
- triptofan (tačka topljenja 282°C) - sadrži azota 13,37 % m/m;
7) natrijum-hidroksid 33% (m/m);
8) reagensi za precipitaciju žive:
a) rastvor natrijum-tiosulfata: rastvori se 80 g natrijum-tiosulfata (Na2S2O3 5H2O) u 1 000 ml vode;
b) natrijum ili kalijum-hipofosfit;
9) kiseline koje se primenjuju za sakupljanje destilata:
- sumporna kiselina, c (1/2 H2SO4) = 0,1 mol/l ili 0,25 mol/l;
- borna kiselina, 40 g/l;
10) standardni volumetrijski rastvori za titraciju:
- natrijum-hidroksid, c (NaOH) = 0,1 mol/l ili 0,25 mol/l;
- sumporna kiselina, c (1/2 H2SO4) = 0,1 mol/l ili 0,25 mol/l;
11) smeša indikatora: rastvori se 2 g metil-crvenog i 1 g metilen-plavog u 1 000 ml 95 %-nog etanola V/V);
12) lakmus-papir;
13) kamenčići za ključanje ili staklene kuglice, prečnika 5 do 7 mm.
Aparatura i pribor
Pored uobičajene laboratorijske opreme, potreban je i sledeći pribor:
1) analitička vaga;
2) aparat za spaljivanje;
3) aparat za destilaciju i titraciju;
4) odmerne tikvice, zapremine 100 ml;
5) Erlenmajer-tikvice, zapremine 250 ml;
6) birete;
7) pipete.
Pripremanje uzorka
Odmeri se 0,5 do 2 g uzorka (najbolje oko 1 g), sa tačnošću 0,001 g, tako da sadržaj azota bude od 0,005 do 0,08 g. Kad je uzorak nehomogen, uzima se veća količina uzorka nego što je predviđeno i, ako pređe 0,08 g azota, uzima se veća količina sumporne kiseline za prihvatanje destilata.
Određivanje
Odmereni uzorak se kvantitativno prenese u kjeldal-bocu zapremine 800 ml i doda 10 g kalijum-sulfata (što je dovoljno) ako se kao katalizator koristi živa ili živin oksid ili 15 g kalijum-sulfata ako se kao katalizator koristi bakar-oksid ili bakar-sulfat.
Doda se odgovarajuća količina katalizatora: 0,65 g (jedna kap) žive ili 0,7 g živinog oksida za sve proizvode. Umesto žive ili živinog oksida može se dodati 0,3 g bakar-oksida ili 0,9 do 1,2 g bakar-sulfata pentrahidrata. Vreme digestije se znatno produžava pri korišćenju bakarnih soli, a živa ima prednost kao katalizator kad je sadržaj proteina veći.
Posle toga se doda 25 ml koncentrovane kiseline za prvi gram suve materije uzorka i 6 do 12 ml za svaki sledeći gram suve materije (za razlaganje skroba i masti potrebno je 6 do 12 ml). Dobro se promućka kako bi se uzorak kompletno navlažio. Zatim se postepeno zagreva kako bi se izbeglo penjenje. Može se dodati i sredstvo protiv penjanja, kao što je parafinski vosak. Pri zagrevanju, povremeno se promućka, dok se uzorak ne ugljeniše i dok ne iščezne para, a zatim se jače zagreva dok tečnost ne proključa. Zagrevanje je zadovoljavajuće ako se kiselina koja ključa kondenzuje oko polovine grla kjeldal-boce, koja treba da se postavi tako da sa vertikalom čini ugao od 30 do 45 °. Kad tečnost postane bistra, zagrevanje se produži još 1h, ako se kao katalizator primeni živa, ili 2 h, ako je jedan katalizator bakar. Zatim se uzorak hladi i ako se pri tom razloženi uzorak iskristališe, za razlaganje se koristi veća količina kiseline nego što je navedeno.
U razloženi uzorak doda se 250 do 350 ml vode kako bi se sulfati potpuno rastvorili, promućka se i ostavi da se ohladi. Pipetom se odmeri 25 ml 0,1 (1/2 H2SO4) mol/l ili 0,25 (1/2 H2SO4) mol/l rastvora sumporne kiseline, zavisno od očekivane količine azota, doda se 100 do 150 ml vode i nekoliko kapi mešanog indikatora. Kraj hladnjaka se uroni u tečnost najmanje 1 cm i polako doda 100 ml 35 %-nog natrijum-hidroksida u kjeldal-bocu.
Napomena. |
Ako je pri digestiji korišćeno više kiseline, proporcionalno se poveća i količina natrijum-hidroksida. |
Ako se kao katalizator koristi živa ili živin oksid, natrijum-hidroksid se pre dodavanja u bocu meša sa 25 ml natrijum-tiosulfata. Ako bi se natrijum-tiosulfat dodao posebno, tiosulfat reaguje sa kiselinom u boci i stvara vodonik-sulfid, što daje pogrešne rezultate. Umesto tiosulfata može se koristiti hipofosfit, pri čemu ne dolazi do stvaranja vodonik-sulfida. Posle dodavanja vode spaljenom uzorku, a pre dodavanja natrijum-hidroksida, doda se 1 g hipofosfita u čvrstom obliku, što je dovoljno za taloženje 1 g žive.
Boca sa uzorkom odmah se spoji sa destilacionim aparatom i zagreva tako da 150 ml destilata pređe za 30 min. Pri kraju tog procesa proveri se reakcija destilata lakmus-papirom. Ako je reakcija alkalna, vreme destilacije se produži. Kraj kondenzatora se izvadi iz tečnosti pre nego što se prekine destilacija da kondenzator ne bi povukao tečnost.
Ako tečnost u toku destilacije u prihvatnom sudu postane alkalna, određivanje se ponovi uz dodavanje odgovarajuće količine kiseline.
Alternativno, destilat se može sakupljati u 100 ml ili 250 ml borne kiseline.
Titracija
Ako se za sakupljanje destilata koristi sumporna kiselina, višak kiseline se titrira 0,1 mol (NaOH)/l ili 0,25 mol (NaOH)/l, do promene boje od ljubičaste u zelenu.
Ako se za sakupljanje destilata koristi borna kiselina, amonijak se titra sa 0,1 mol (1/2 H2SO4)/l ili 0,25 mol (1/2 H2SO4)/l sumpornom kiselinom dok se boja ne promeni iz zelene u ljubičastu. Preporučuje se istovremena titracija amonijaka u toku destilacije jer olakšava određivanje krajnje tačke. Krajnja tačka pokazuje promenu boje smeše indikatora.
Ako nije mogućna uporedna titracija, treba je izvesti neposredno posle destilacije, vodeći računa o tome da temperatura destilata ne bude viša od 25°C. Pod tim uslovima izbegava se gubitak amonijaka.
Slepa proba
Slepa proba se napravi sa 1 g saharoze.
Kontrolna proba
Kontrolna proba se priprema tako što se odredi sadržaj azota u acetanilidu ili triptofanu, uz dodavanje 1 g saharoze.
Supstancija za kontrolnu probu bira se u odnosu na brzinu razlaganja uzorka koji se analizira. Acetanilid se lako razloži, dok je razlaganje triptofana mnogo teže. Triptofan se suši pre korišćenja.
Izračunavanje
1. Izračunavanje sadržaja azota
Ako je destilat hvatan u sumpornoj kiselini, a količina sumporne kiseline koja je korišćena za hvatanje amonijaka u uzorku ista kao i za slepu probu, sadržaj azota u procentima je:
(V0 - V1) · T · 0,014 · 100 |
= |
1,4 (V0 - V1) · T |
||
m |
m |
gde je:
V0 - broj mililitara natrijum-hidroksida utrošenog za slepu probu;
V1 - broj mililitara natrijum-hidroksida utrošenog pri određivanju;
T - molaritet rastvora natrijum-hidroksida koji se koristi za titraciju;
m - masa uzorka, u gramima.
Ako je destilat hvatan u bornoj kiselini, sadržaj azota se izražava u procentima mase, i to:
(V1 - V0) · T · 0,014 · 100 |
= |
1,4 (V1 - V0) · T |
||
m |
m |
gde je:
V0 - broj mililitara sumporne kiseline utrošene za slepu probu;
V1 - broj mililitara sumporne kiseline utrošene za određivanje;
T - molaritet rastvora sumporne kiseline koji se koristi za titraciju;
m - masa uzorka, u gramima.
Kao rezultat uzima se aritmetička sredina iz dva uporedna određivanja, ako su ispunjeni zahtevi u pogledu ponovljivosti. Rezultat se daje najmanje na 0,01 % (m/m).
2. Sadržaj sirovih proteina dobija se množenjem sadržaja azota faktorom 6,25.
Ponovljivost
Razlika između rezultata dva određivanja sadržaja azota koja je istovremeno ili neposredno jedno za drugim izvršio isti analitičar ne sme preći 0,03 % apsolutne vrednosti kad je sadržaj azota do 3 % (m/m) ili 1% relativne srednje vrednosti kad sadržaj azota iznosi od 3 do 6 % i 0,06 apsolutne vrednosti za sadržaj azota veći od 6 % (m/m).
Napomena. |
Za određivanje sadržaja sirovih proteina mogu se koristiti automatski ili poluautomatski uređaji koji rade na principu Kjeldalove metode. |
8. Određivanje amonijačnog azota
Primena
Metoda se primenjuje za određivanje amonijačnog azota u stočnoj hrani.
Pribor
Pored uobičajene laboratorijske opreme, potreban je i sledeći pribor:
1) kjeldal-tikvica, zapremine 750 ml;
2) aparat za destilaciju, sa potrebnim delovima;
3) bireta, zapremine 50 ml;
4) konična tikvica (erlenmajer).
Reagensi
Za određivanje amonijačnog azota, koriste se sledeći reagensi:
1) magnezijum-oksid, MgO;
2) rastvor sumporne kiseline, c(1/2H2SO4) = 0,1 mol/l;
3) 0,5 %-ni rastvor metil-crvenog u 96 %-nom etanolu.
Određivanje
Izmeri se 5 do 10 g uzorka i prenese u bocu za destilaciju, zapremine 750 ml, u koju se doda 250 ml destilovane vode i 3 g MgO. Destilaciona boca se preko staklene destilacione kruške poveže s hladnjakom i koničnom tikvicom u koju je biretom prethodno uneto 50 ml 0,1 mol (H2SO4)/l i dodato metil-crvenog. Blagim zagrevanjem započne destilacija, koja traje dok se u koničnoj tikvici ne predestiliše oko 150 ml destilata ili dok se lakmusom ne utvrdi da je destilacija azota završena.
Na kraju se destilat, uhvaćen u 0,1 mol (H2SO4)/l, titrira sa 0,1 mol (NaOH)/l i količina predestilisanog azota izračunava se na isti način kao i pri određivanju ukupnog azota.
Izračunavanje
Sadržaj amonijačnog azota izražava se u procentima mase uzetog uzorka (vazduhom sušenog) i izračunava po formuli:
X = |
(a - b) · 0,0014 · 100 |
% |
m |
gde je:
a - količina 0,1 mol (1/2H2SO4)/l, upotrebljena na početku analize u koničnoj tikvici, u mililitrima;
b - količina 0,1 mol (NaOH) utrošena pri titraciji za neutralisanje slobodne sumporne kiseline, u mililitrima;
0,0014 - količina azota koja odgovara količini od 1 ml 0,1 mol (1/2H2SO4)/l;
m - masa izmerenog uzorka, u gramima.
9. Određivanje uree - spektrofotometrijski
Princip i primena
Određivanje sadržaja uree utvrđuje se spektrofotometrijskim merenjem apsorbancije na 420 nm i primenjuje se za određivanje uree u krmnim smešama i sirovinama za izradu krmnih smeša.
Aparati i pribor
Pored uobičajene laboratorijske opreme, koristi se i sledeći pribor:
1) pipete;
2) odmerne tikvice, zapremine 100 ml, 250 ml, 500 ml i 1 000 ml;
3) vodeno kupatilo;
4) spektrofotometar.
Reagensi
Za određivanje uree koriste se sledeći reagensi:
1) p-dimetil-aminobenzaldehid (DMAB): rastvori se 16 g reagensa DMAB u 1 l alkohola i doda 100 ml hlorovodonične kiseline (J20 = 1,19 g). Rastvor je stabilan mesec dana. Izradom novog reagensa, priprema se nova baždarna kriva;
2) rastvor cink-acetata: rastvori se 22 g (CH3COC) Zn · 2 H2O u vodi, doda 3 ml sirćetne kiseline i dopuni vodom do 100 ml;
3) rastvor kalijum-ferocijanida: u vodi se rastvori 10,6 g K4Fe (CN)6 · 3 H2O i dopuni vodom do 100 ml;
4) aktivni ugalj;
5) fosfatni pufer, PH7: rastvori se 3,403 g bezvodnog KH2PO4 i 4,355 g bezvodnog K2HPO4 posebno, u po 100 ml sveže destilovane vode. Ta dva rastvora se pomešaju i dopune do 1 l destilovanom vodom;
6) urea - standardni rastvor;
a) osnovni rastvor 5 mg/ml: rastvori se 5 g (mereno sa tačnošću 0,001 g) uree u 1 l destilovane vode;
b) radni rastvor: pipetira se 2, 4, 6, 8, 10, 12, 14, 16, 18 i 20 ml osnovnog rastvora u sudove zapremine 250 ml i dopuni do crte fosfatnim puferom PH7. Ti rastvori sadrže 0,2, 0,4, 0,6, 0,8, 1,0, 1,2, 1,4, 1,8 i 2,0 mg uree/5 ml;
v) referentni standard: kao referentni standard koristi se radni rastvor sa koncentracijom uree 1,0 mg/l ml. Rastvor je stabilan nedelju dana na temperaturi nižoj od 24°C.
Pripremanje standardne krive
U epruvete se otpipetira po 5 ml radnih rastvora uree i u svaku epruvetu se doda po 5 ml rastvora DMAB. Slepa proba priprema se sa 5 ml fosfatnog pufera i 5 ml rastvora DMAB. Svaka proba se pomeša i ostavi 10 minuta u vodenom kupatilu na temperaturi 25°C. Očita se apsorbancija na spektrofotometru, na talasnoj dužini 420 nm, i slepom probom odredi nula apsorbancije, a zatim se konstruiše baždarna kriva.
Određivanje
U odmernu tikvicu zapremine 500 ml odmeri se 1 g uzorka, doda 1 g aktivnog uglja, 250 ml destilovane vode, 5 ml rastvora cink-acetata i 5 ml rastvora kalijum-ferocijanida. Mućka se 30 min i do 500 ml dopuni destilovanom vodom. Promućka se i ostavi da se slegne. Kroz filtrir-papir (NO 40) rastvor se profiltrira i za dalju analizu se uzima čist filtrat. Po 5 ml filtrata pipetira se u epruvete, doda 5 ml DMAB i odmah pomeša. U svaku seriju uzorka uključi se referentni standard (5 ml referentnog standarda i 5 ml rastvora DMAB) i slepa proba. Svi rastvori se ostave 10 min u vodenom kupatilu na temperaturi 25°C. Posle toga se očita apsorbancija na spektrofotometru, na talasnoj dužini od 420 nm, uz slepu probu.
Izračunavanje
Sadržaj uree izražava se u procentima i izračunava se po sledećoj formuli:
urea = |
1,0 · apsorbancija uzorka |
|
apsorbancija standarda · mg uzorka u alikvotu |
Primena
Određivanje kazeina vrši se u mleku u prahu kad se to mleko koristi kao stočna hrana.
Određivanje
Odmeri se 10 g uzorka i rastvori sa 140 ml destilovane vode u čaši zapremine 200 ml. Voda se dodaje postepeno da se ne bi stvarale grudvice. Tako pripremljen uzorak zagreva se do 40°C, uz stalno mešanje i kontrolu termometrom. Kad uzorak dostigne temperaturu 40°C, dodaje se 1 ml zasićenog rastvora stipse K2Al2(SO4)2 · 24H2O radi izdvajanja taloga. Ako se talog ne izdvoji, treba dodati, kap po kap, još 0,5 ml rastvora stipse. Posle toga se sadržaj iz čaše filtrira, pa se čaša i sadržaj na filtrir-papiru tri puta ispiraju destilovanom vodom.
Filtrir-papir, sa sadržajem, prenese se u kjeldal-bocu i postupa se kao i pri određivanju azota.
Izračunavanje
Sadržaj kazeina izračunava se tako što se utvrđena količina azota pomnoži sa 6,38 i dobija procent od mase uzorka.
Primena
Određivanje laktoalbumina vrši se u proizvodima mleka za stočnu hranu.
Određivanje
Za određivanje laktoalbumina koristi se filtrat posle odvajanja kazeina, što je utvrđeno u metodi određivanja kazeina. Dobijeni filtrat najpre se neutrališe 10%-nim NaOH, a zatim se doda 0,3 ml koncentrovane sirćetne kiseline razblažene vodom u odnosu 1:9. Tako dobijena neutralna mešavina zagreva se na vodenom kupatilu dok se sav laktoalbumin ne istaloži. Dobijeni talog se sakupi na filtru koji je ispran kiselinom, ispira destilovanom vodom, a zatim se određuje direktno azot, što je opisano u metodi određivanja azota i sirovih proteina propisanoj u ovom pravilniku.
Izračunavanje
Sadržaj laktoalbumina izračunava se tako što se sadržaj azota množi koeficijentom 6,38.
Princip i primena
Metoda se primenjuje pri određivanju sirovih masti u stočnoj hrani, osim u uljaricama i sporednim proizvodima od uljarica. Za to određivanje primenjuju se dva postupka, zavisno od vrste stočne hrane. Postupak A (direktna ekstrakcija dietil-etrom) primenjuje se za sve vrste stočne hrane koje se ne određuju postupkom B. Postupak B primenjuje se za ispitivanje stočne hrane iz koje nije mogućna direktna ekstrakcija dietil-etrom bez prethodne pripreme stočne hrane životinjskog porekla, glutena, suve kaše od krompira, suvog kvasca, delova biskvita i jela, proizvoda od mleka i stočne hrane koja sadrži najmanje 40% pomenutih sirovina.
Za proizvode sa visokim sadržajem ulja i masti ili visokim sadržajem vlage, koji se teško isitnjavaju, neop-hodno je da se uzorak prethodno tretira.
Princip metode A zasniva se na ekstrakciji dietil-etrom, odstranjenju rastvarača destilacijom, sušenju i merenju ostatka.
Princip metode B zasniva se na hidrolizi uzorka hlorovodoničnom kiselinom, hlađenju i filtraciji rastvora, a zatim u ispiranju i sušenju ostatka dobijenog ekstrakcijom sa dietil-etrom.
Reagensi i pomoćna sredstva
Za određivanje sirovih masti koriste se sledeći reagensi:
1) dietil-etar, bezvodni, slobodan od peroksida (r20 = 0,720 g/ml, tačke ključanja 34 do 35°C);
2) ugljen-tetrahlorid;
3) natrijum-sulfat, bezvodni;
4) hlorovodonična kiselina, C(HCl) = 3 mol/l;
5) sredstvo za filtraciju.
Pribor
Pored ostale laboratorijske opreme, koristi se sledeći pribor:
1) sohlet-ekstraktor ili slična aparatura;
2) aparat za zagrevanje, sa kontrolom temperature;
3) vakuumska sušnica (pritisak ne manji od 13 kPa);
4) sušnica sa kontrolom temperature od 95 do 98°C;
5) analitička vaga;
6) konična tikvica (erlenmajer), zapremine 300 ml ili stakleni pehar zapremine 400 ml.
Određivanje
Postupak A
Odmeri se 5 g, uzorka za ispitivanje, sa tačnošću 0,001 g pomeša sa 2 do 3 g natrijum-sulfata i smeša se prenese u čahuru ekstraktora koja se prekrije pamukom. Čahura se prenese u eksikator i ekstrahuje 6 h dietil-etrom. Pri korišćenju sohlet-ekstraktora reguliše se temperatura tako da se postigne 15 sifoniranja na sat. Zatim se sakupi suvi ostatak i odmeri tikvica u kojoj se nalaze i delovi kamena plovućca. Potom se izvrši destilacija i ostatak suši u toku 1,5 h u vakuumskoj sušnici pri temperaturi 75°C, hladi u eksikatoru i meri sa tačnošću 0,001 g. Ponovo se suši 30 min, do konstantne mase.
Postupak B
Odmeri se 2,5 g uzorka za ispitivanje, sa tačnošću 0,001 g, i prenese u koničnu tikvicu ili staklenu čašu. Za proizvode sa niskim sadržajem ekstrakta dietila uzorak za ispitivanje iznosi 5 g.
U koničnu tikvicu sa uzorkom za ispitivanje doda se 100 ml hlorovodonične kiseline i delovi kamena plovućca. Konična tikvica se pokrije sahatnim staklom i prenese na aparat za zagrevanje, gde lagano ključa 1 h. Zatim se hladi i dodaje mala količina sredstva za filtriranje kako bi se za vreme filtracije sprečio gubitak ulja ili masti. Filtracija se obavlja kroz dupli filtrir-papir, a ostatak ispere hladnom vodom dok ne prestane kisela reakcija. Proveri se da li na površini filtrata ima ulja ili masti. Ako se utvrdi prisustvo ulja ili masti, uzorak se mora ekstrahovati dietil-etrom, kao što je opisano u postupku A, pre hidrolize.
Dupli filtrir-papir se sa ostatkom prenese na sahatno staklo i suši 1,5 h u peći na temperaturi 95 do 98°C, a zatim se prenese u čahuru ekstraktora i 6 h ekstrahuje sa dietil-etrom. Dalje se postupa kao što je utvrđeno postupkom A.
Izračunavanje
Ekstrakt dietil-etra izražava se kao procent mase uzorka i izračunava se po sledećoj formuli:
(m1 - m2) |
· 100 |
||
m0 |
gde je:
m0 - masa uzorka za ispitivanje, u gramima;
m1 - masa konične tikvice sa delovima kamena plovućca i suvim ekstraktom etra;
m2 - masa konične tikvice sa delovima kamena plovućca, u gramima.
Rezultat se izražava jednom decimalom.
Ponovljivost
Razlika između dva paralelna određivanja koja je istovremeno ili jedno za drugim izveo isti analitičar ne sme biti veća od 0,3 % ulja ili masti.
13. Određivanje slobodnih masnih kiselina
Princip i primena
Metoda se primenjuje pri ispitivanju stočne hrane siromašne skrobom i komponenata stočne hrane bogate mastima. Metodom se određuju slobodne masne kiseline izražene kao oleinska kiselina.
Princip se zasniva na titraciji uzorka ekstrakta etara natrijum-hidroksidom.
Reagensi
Za određivanje slobodnih masnih kiselina koriste se sledeći reagensi:
1) rastvor c(NaOH) = 0,1 mol/l (ili KOH) u 96 %-nom etanolu;
2) smeša etanola i dietil-etra 1 + 1 (V/V), neutralisana;
3) fenolftalein, 1 %-ni rastvor u etanolu.
Pribor
Koristi se vodeno kupatilo.
Određivanje
Posle ekstrakcije u dietil-etru, ostatak masti se otopi u 100 ml smeše etanola i deitil-etra 1 + 1 (V/V). Ako se taj ostatak slabo otapa, otapanje se ubrzava grejanjem rastvora u boci na vodenom kupatilu. Rastvor se dobro promućka, zatim se doda dva do tri kapi fenolftaleina kao indikatora i titrira natrijum-hidroksidom.
Izračunavanje
Jedan mililitar rastvora c (NaOH) = 0,1 mol/l odgovara 0,0285 g oleinske kiseline (C18H34O2). Sadržaj slobodnih masnih kiselina, kao oleinska kiselina u procentima mase, izračunava se prema formuli:
C · 0,0285 · 100 |
||
odvaga masti, u gramima |
gde je:
C - broj mililitara 0,1 mol/l NaOH.
14. Određivanje kiselinskog stepena
Princip i primena
Princip se zasniva na ekstrakciji uzorka 67 %-nim etanolom i titraciji sa 0,1 mol/l NaOH. Metoda se primenjuje za određivanje slobodnih masnih kiselina (kiselinskog stepena) u stočnoj hrani bogatoj skrobom.
Reagensi
Za određivanje kiselinskog stepena koriste se sledeći reagensi, čistoće pro analysi:
1) etanol, 67 %-ni (V/V) = 0,893 g/ml neutralisan natrijum-hidroksidom uz fenolftalein kao indikator;
2) indikator, 3 %-ni rastvor fenolftaleina u 70 %-nom etanolu;
3) rastvor c (NaOH) = 0,1 mol/l NaOH poznatog faktora.
Pribor
Za određivanje kiselinskog stepena koristi se sledeći pribor:
1) analitička vaga;
2) erlenmajer-tikvica;
3) pipeta;
4) električna mućkalica;
5) filtrir-papir;
6) mikrobireta po Bangu, podela 1/100.
Pripremanje uzorka
Uzorak se mora isitniti tako da najmanje 90 % čestica prolazi kroz sito veličine otvora 1 mm.
Određivanje
Izmeri se 10 g isitnjenog uzorka stočne hrane, sa tačnošću od 0,01 g, i prenese u erlenmajer-tikvicu. Zatim se pipetom doda 50 ml 67 %-nog etanola i kružnim kretanjem mućka 15 min pri temperaturi 20°C. Ako se koristi električna mućkalica, dovoljno je 5 min mirovanja da se talog slegne, a rastvor se procedi kroz nabrani filtrir-papir.
Posle 5 min pipetom se uzme 25 ml filtrata i prenese u drugu erlenmajer-tikvicu, zatim se doda 0,5 ml indi-katora fenol-ftaleina i titrira rastvorom 0,1 mol/l NaOH dok se ne pojavi postojana ljubičasta boja.
Izračunavanje
Kiselinski stepen označava broj ml 1 mol/l NaOH potrebnih za neutralizaciju slobodnih masnih kiselina u 100 g uzorka i izračunava se po sledećoj formuli:
kiselinski stepen = 2a
gde je:
a - utrošen broj ml 0,1 mol (NaOH)/l za neutralizaciju.
Ponovljivost
Razlika između rezultata dva određivanja izvršena paralelno ili ubrzo jedno za drugim ne sme iznositi više od 0,2 jedinice za vrednost kiselinskog stepena do 3, a 0,3 jedinice - za vrednost kiselinskog stepena većeg od 3.
Princip i primena
Princip se zasniva na elektrohemijskom merenju pH vrednosti vodenog ekstrakta stočne hrane.
Metoda se primenjuje za sva hraniva (osim masti, ulja i sl.) i mešanu stočnu hranu. Ako postoji osnovana sumnja na homogenost, uzima se odgovarajući veći uzorak (npr. za silažu 100 g u 1 000 ml).
Pribor
Osim uobičajene laboratorijske opreme, potrebno je pripremiti:
1) pH - metar sa staklenom i Kalomelovom elektrodom;
2) mućkalica sa oko 40 O/min i posudama odgovarajuće zapremine.
Određivanje
Odmeri se 10 g uzorka i sa 100 ml vode prenese u sud za ekstrakciju. Radi bolje ekstrakcije, potrebno je dodati nekoliko staklenih perlica. Uzorak se ekstrahuje na mućkalici 15 min. Sadržaj se procedi i u bistrom filtaratu se odredi pH vrednost.
16. Određivanje sirove celuloze
Princip i primena
Metoda se zasniva na tretiranju uzorka sumpornom kiselinom i kalijum-hidroksidom, pa se ostatak odvoji filtriranjem i, posle sušenja i žarenja, izmeri.
Pribor
Pored uobičajene laboratorijske opreme, potreban je i sledeći pribor:
1) analitička vaga;
2) izvori zagrevanja (rešo ili plamenik);
3) kvarcni filtrirni lončići i oprema za vakuumsko filtriranje;
4) mufolna peć (peć za žarenje);
5) mlin sa sitom.
Reagensi i pomoćna sredstva
Za određivanje sirove celuloze potrebni su sledeći reagensi čistoće pro analysi i destilovana voda:
1) rastvori sumporne kiseline c(1/2H2SO4) = 0,26 mol/l;
2) rastvori kalijum-hidroksida c(KOH) = 0,23 mol/l;
3) sredstva protiv penušanja (npr. silikon);
4) aceton;
5) dietil-etar, čist;
6) rastvor hlorovodonične kiseline c(HCl) = 0,5 mol/l.
Kvarcni pesak, ispran i žaren na temperaturi od 550°C ± 25°C, kuva se 30 min u 4 mol/l HCl. Zatim se ispira vodom, dok se ne oslobodi od hlorida, i žari na temperaturi 550 ± 25°C do konstantne mase.
Pripremanje uzorka
Ako uzorak sadrži više od 10% masti, prethodno se podvrgava odmašćivanju ekstrakcijom u dietil-etru. U guč-lončić sa filtrir-papirom na dnu odmeri se 3 g uzorka za ispitivanje i tri puta ispira sa po 50 ml dietil-etra, koji se odsisiva pomoću vakuuma. Odmašćen uzorak se kvantitativno prenese u čašu zapremine 600 ml i određuje sirova celuloza.
Za hranu iz koje se masti ne mogu direktno ekstrahovati u dietil-etru, uzorak se kuva sa 1,25 mol/l sumpornom kiselinom, zatim tri puta ispira acetonom (oko 100 ml) i tri puta sa po 50 ml dietil-etra. Ostatak se kvantitativno prenese u čašu i nastavi se kuvanje sa kalijum-hidroksidom. To se primenjuje na hranu "zamenu mleka" i ostatke pekarske industrije.
Od hrane koja sadrži više od 2% kalcijuma odmeri se 3 g uzorka i tretira 5 min sa 100 ml hladnog rastvora hlorovodonične kiseline, koncentracije 0,5 mol/l. Ostatak se filtrira i ispira hladnom vodom. Koristi se za određivanje sirove celuloze.
Određivanje
U visoku čašu sa ravnim rubom zapremine 600 ml, odmeri se 3 g uzorka sa tačnošću 0,001 g, i doda se 200 ml sumporne kiseline. Čaša se stavi iznad izvora toplote i greje da bi sadržaj što pre proključao. Ključanje se zatim produžava još 30 min. Da voda pri tom ne bi isparila, odnosno da se ne bi povećala koncentracija kiseline u posudi sa uzorkom, na čaše se stavljaju boce okruglog dna kroz koje protiče hladna voda, tako da one služe kao kondenzatori. Umesto tih boca mogu se upotrebiti i druge vrste hladnjaka koji sprečavaju isparavanje tečnosti. Posle 30 min ključanja, dodaje se 50 ml hladne vode i sadržaj se filtrira pod vakuumom preko guč-lončića sa pripremljenom kvarcnom masom. Kad se filtracija završi, pristupa se ispiranju preostale kiseline iz ostatka uzorka u lončiću sa vrućom destilovanom vodom (95 - 100°C) dok filtrat ne postane neutralan.
Nerastvoreni materijal se pomoću staklenog štapića prenese iz lončića u već upotrebljenu čašu i doda se 200 ml rastvora kalijum-hidroksida. Čaša se stavi na izvor toplote i greje do ključanja, koje traje 30 min. Zatim se doda 50 ml hladne destilovane vode i filtrira preko upotrebljenog guč-lončića. Ostatak se ispira vrućom destilovanom vodom sve dok se ne otklone tragovi kalijum-hidroksida. Zatim se ispiranje nastavi na taj način što se aceton dodaje tri puta, u ukupnoj količini od približno 50 ml, i, na kraju se ispiranje dietil-etrom u ukupnoj količini od 50 ml, ponovi tri puta.
Ostatak nerastvorenog uzorka (sirova celuloza) suši se u guč-lončiću na temperaturi 105°C, a zatim se hladi u eksikatoru i meri do konstantne mase - m1 (masa 1). Guč-lončić se stavi u mufolnu peć koja je zagrejana na temperaturu od 550 do 600°C. Posle žarenja hladi se u eksikatoru i meri m2 (masa 2).
Izračunavanje
Sadržaj sirove celuloze izražava se u procentima mase i izračunava po sledećoj formuli:
(m1 - m2) |
· 100 |
||
m |
gde je:
m - masa uzetog uzorka, u gramima;
m1 - masa lončića sa celulozom, u gramima;
m2 - masa praznog lončića, u gramima.
Kao rezultat uzima se srednja aritmetička vrednost najmanje dva uporedna određivanja, ako su ispunjeni zahtevi u pogledu ponovljivosti.
Rezultat se zaokrugli na dve decimale.
Ponovljivost
Razlika između rezultata dva uporedna određivanja, koja istovremeno ili ubrzo jedno za drugim izvodi isti analitičar, ne sme biti veća od 0,5%.
Princip i primena
Sadržaj skroba u uzorku delimično se razloži hlorovodoničnom kiselinom do dekstrina i glukoze. Posle otklanjanja proteina iz smeše odredi se optička aktivnost filtriranog rastvora. Posle korekcije rezultata za optičku aktivnost optički aktivnih supstancija koje nisu skrob, dekstrin i glukoza, izračunava se sadržaj skroba pomoću formule.
Metoda se primenjuje pri ispitivanju stočne hrane životinjskog i biljnog porekla, osim kad su u pitanju smeše koje sadrže krompir, kašu od repe, rezance od repe ili celu repu, kao i proizvodi sa visokim sadržajem insulina.
Reagensi
Svi reagensi moraju biti čistoće pro analysi. Za pripremanje rastvora koristi se redestilovana voda.
Koriste se sledeći reagensi:
1) etanol 40%-ni (V/V) r = 0,948 g/ml), neutralan u odnosu na fenolftalein kao indikator;
2) metil-crveno, indikator 1 g/l etanola 96%-nog (V/V);
3) hlorovodonična kiselina 25%-na (r = 1,126 g/ml);
4) hlorovodonična kiselina 1,128%-na (m/V);
5) Carrez I: rastvori se 21,9 g Zn (CH3COO)2 · 2H2O i 3 g CH3COOH u vodi i dopuni vodom do 1 000 ml;
6) Carrez II: rastvori se 10,6 g K4Fe (CN)6 · 3H2O u vodi i dopuni vodom do 100 ml;
7) aktivni ugalj (carbo medicinalis).
Pribor
Pored uobičajene laboratorijske opreme, koristi se i sledeći pribor:
1) mlin za fino mlevenje;
2) analitička vaga;
3) vodeno kupatilo, 100°C;
4) tikvica, zapremina 100 ml;
5) menzura, zapremine 100 ml;
6) pipeta, zapremine 50 ml;
7) pipeta, zapremine 25 ml;
8) pipeta, zapremine 10 ml;
9) konična boca, zapremine 200 ml;
10) polarimetar ili saharimetar sa polarimetarskim cevima, dužine 200 mm.
Pripremanje uzorka za analizu
Prvih nekoliko grama u mlinu samlevenog uzorka se odbaci, a ostatak se samelje tako da čestice mliva prolaze kroz sito veličine otvora 0,5 mm. Samleveni uzorak mora biti dobro homogenizovan kako bi se izbeglo izdvajanje pojedinih frakcija. Samleveni uzorak drži se u dobro zatvorenoj boci i analizira što je mogućno pre.
Određivanje
Određivanje ukupne optičke aktivnosti
Izmeri sa 2,5 g uzorka, sa tačnošću od 0,001 g, i sa 25 ml 1,128 % HCl prenese se u odmernu tikvicu zapremine 100 ml. Sadržaj se dobro izmućka (da ne ostanu gromuljice), grlić tikvice se ispere sa novih 25 ml HCl, a sadržaj se još jedanput dobro promućka i tikvica zaroni u ključalo vodeno kupatilo. Količina vode u kupatilu mora biti tolika da posle uranjanja tikvice ključanje ne prestane.
U toku prva tri minuta sadržaj se snažno promućka nekoliko puta, ali se tikvica ne vadi iz kupatila. Posle tačno 15 min tikvica se izvadi iz vodenog kupatila, doda se oko 30 ml hladne redestilovane vode i sadržaj u tikvici ohladi se vodom do sobne temperature.
Za taloženje proteina doda se 5 ml Carreza I, mućka se 1 min doda 5 ml Carreza II, ponovo mućka 1 min i dopuni vodom do 100 ml, promućka i filtrira.
Sadržaj tikvice se dobro promućka, ostavi se 15 min i filtrira kroz suv filtrir-papir. Ako filtrat nije bistar, ponovi se određivanje, a za taloženje proteina uzima se 10 ml Carreza I i 10 ml Carreza II.
Ako rastvor i posle toga ne postane bistar, doda se oko 2 g aktivnog uglja, dobro promućka i ponovo filtrira kroz suvi filtrir-papir.
Dobijenim bistrim rastvorom napuni se polarimetarska cev, dužine 200 mm, i odredi optička aktivnost.
Određivanje optičke aktivnosti supstancija rastvorljivih u etanolu 40 % (V/V)
Izmeri se 5 g uzorka, sa tačnošću od 0,001 g i sa 80 ml etanola 40 % (V/V) prenese u odmernu tikvicu zapremine 100 ml, dobro promućka 1 h i ostavi na sobnoj tempreturi. Za to vreme sadržaj tikvice se svakih 10 min promućka, zatim dopuni etanolom do oznake, ponovo promućka i filtrira.
Napomena. |
Kad stočna hrana sadrži više od 5% laktoze, posle odmeravanja uzorka i dodavanja 80 ml etalona sadržaj se prenese u odmernu tikvicu zapremine 100 ml, koja se zatim spoji sa hladnjakom i drži 30 min na vodenom kupatilu na temperaturi 50°C. Posle toga sadržaj se hladi, dopuni do oznake i filtrira. |
Odmeri se 50 ml filtrata, stavi u tikvicu zapremine 250 ml, doda 2 ml 25%-ne HCl, spoji sa hladnjakom i zaroni u ključalo vodeno kupatilo i drži tačno 15 min. Zatim se sadržaj preruči u odmernu tikvicu zapremine 100 ml, tikvica se ispere hladnom vodom i sadržaj ohladi vodom do sobne temperature.
Za taloženje proteina doda se 5 ml Carreza I, mućka se 1 min, doda 5 ml Carreza II, ponovo mućka 1 min, dopuni vodom do 100 ml, promućka i filtrira.
Polarizaciona cev dužine 200 mm napuni se bistrim rastvorom i odredi se optička aktivnost.
Merenje se izvodi na polarimetru ili saharimetru, na sobnoj temperaturi.
Napomena. |
Kad su u pitanju hraniva kao što je koštano brašno, mesno brašno, riblje brašno, alkalno tretirana brašna i brašna sa visokim sadržajem kreča, obavlja se neutralizacija. |
Izračunavanje
Sadržaj skroba izražava se u procentima i izračunava po sledećoj formuli:
W = 10 000 |
a |
|
[a]D20 · l · c |
gde je:
a = P - p;
P - ukupna optička aktivnost, u kružnim stepenima;
p - optička aktivnost frakcije rastvorene u etanolu 40% (V/V), u kružnim stepenima;
[a]D20 - specifični ugao rotacije dat u tabeli;
l - dužina polarimetarske cevi;
c - konstanta čija vrednost zavisi od tipa polarimetra;
c = 1 , za polarimetar sa kružnom skalom;
c = 2,8885 za polarimetar sa internacionalnom skalom za šećer;
c = 2,8854, za Venckeov (Ventzke) tip polarimetra.
Vrednosti za specifične optičke rotacije nekoliko vrsta skroba određenih ovom metodom u kružnim stepenima date su u tabeli.
Skrob |
Specifična optička
rotacija |
Pšenica |
182,7 |
Ako je odvaga iznosila 2,5 g i dužina polarimetarske cevi 200 mm, a za specifičnu rotaciju uzeta prosečna vrednost od 185,05, sadržaj skroba u procentima iznosi:
W% = 10,887 - za kružnu skalu;
W% = 3,769 - za skalu za šećer;
W% = 3,773 - za Venckeovu (Ventzke) skalu.
Kao rezultat jednog određivanja uzima se artimetička sredina pet očitavanja optičke rotacije jednog rastvora. Za konačan rezultat uzorka uzima se aritmetička sredina najmanje dva uporedna određivanja izvršena istovremeno na istom uzorku.
Ponovljivost
Razlika između dva paralelna određivanja na istom uzorku ne sme biti veća od:
- 0,4%, u apsolutnoj vrednosti - za sadržaj do 40% skroba;
- 1,0%, u relativnoj vrednosti - za sadržaj skroba veći od 40%.
18. Određivanje sirovog pepela
Princip i primena
Metoda se zasniva na razaranju uzorka žarenjem na temperaturi 550°C i merenju ostatka, a pepeo se izražava u procentima mase. Metoda se koristi za određivanje pepela u stočnoj hrani.
Pribor
Pored uobičajene laboratorijske opreme, koriste se i:
1) analitička vaga;
2) mufolna peć, sa mogućnošću regulacije temperature na 550°C. Temperatura u peći u blizini posuda za žarenje ne sme se razlikovati za više od 20°C u odnosu na određenu temperaturu;
3) sušnica podešena na temperaturu od 103 ± 2°C;
4) izvori zagrevanja (rešo ili plamenik);
5) posuda za žarenje od porculana, platine ili legure platine i srebra ili drugog materijala pogodnog za uslove ispitivanja, odgovarajućeg oblika, zapremine od 20 do 30 ml i visine do 5 cm.
Napomena. |
Za uzorke koji u toku ugljenisanja imaju tendenciju bubrenja treba koristiti posude zapremine 30 do 250 ml i visine do 10 cm; |
6) eksikator, snabdeven efikasnim sredstvom za sušenje.
Određivanje
U posudu za žarenje koja je prethodno zagrevana 30 min u mufolnoj peći, na temperaturi od 550°C, ohlađenu u eksikatoru i izmerenu sa tačnošću 0,0001 g, odmeri se oko 3 g uzorka za ispitivanje, sa tačnošću 0,0001 g.
Posuda za žarenje sa uzorkom postepeno se zagreva na rešou ili plameniku, do ugljenisanja uzorka, tako da u toku čitavog postupka ne dođe do gubitaka neorganske materije. Posuda se prenese u mufolnu peć, prethodno regulisanu na 550°C, i ostavi u peći 3 h. Vizuelno se proveri da li je pepeo oslobođen ugljenisanih čestica. Ako su se ugljenisane čestice zadržale, posuda se ponovo vraća u mufolnu peć i greje 1 h. Ako su ugljenisane čestice još vidljive ili ako se sumnja u njihovo prisustvo, posuda se ostavi da se pepeo ohladi. Pepeo se zatim navlaži destilovanom vodom, pažljivo osuši u sušnici na temperaturi 103 ±2°C, a zatim se posuda vrati u mufolnu peć i ponovo zagreva 1 h. Posle toga stavi se u eksikator da se ohladi do sobne temperature i brzo meri, sa tačnošću od 0,0001 g.
Napomena. |
Pepeo dobijen prema navedenom postupku može se kasnije koristiti za određivanje pepela nerastvorljivog u hlorovodoničnoj kiselini. |
Izračunavanje
Sirovi pepeo izražava se u procentima mase i izračunava po sledećoj formuli:
(m2 - m0) · |
100 |
|
m1 - m0 |
gde je:
m0 - masa prazne posude, u gramima;
m1 - masa posude sa uzorkom za ispitivanje, u gramima;
m2 - masa posude i sirovog pepela, u gramima.
Kao rezultat se uzima aritmetička srednja vrednost dva paralelna određivanja, s tim da su zadovoljeni uslovi ponovljivosti.
Rezultat se izražava dvema decimalama.
Ponovljivost
Razlika između dva paralelna određivanja istog uzorka ne sme biti veća od:
0,3 jedinice (u apsolutnoj vrednosti) - za sadržaj pepela manji od 3 %;
0,4 jedinice (u apsolutnoj vrednosti) - za sadržaj pepela od 3 do 5 %;
0,5 jedinice (u apsolutnoj vrednosti) - za sadržaj pepela 5 do 20 %;
0,7 jedinice (u apsolutnoj vrednosti) - za sadržaj pepela od 20 do 40 %;
1 jedinice (u apsolutnoj vrednosti) - za sadržaj pepela veći od 40 %.
19. Određivanje pepela nerastvorljivog u hlorovodoničnoj kiselini
Princip i primena
Za određivanje pepela nerastvorljivog u hlorovodoničnoj kiselini primenjuju se dva postupka, zavisno od vrste stočne hrane.
Princip postupka A sastoji se u razaranju organske materije sagorevanjem, a dobijeni pepeo tretira 10 %-nim rastvorom hlorovodonične kiseline, filtrira, suši, spaljuje i dobijeni ostatak meri.
Postupak A primenjuje se na hraniva organskog porekla i na krmne smeše u kojima se pretpostavlja sadržaj pepela do 1 %.
Princip postupka B zasniva se na izdvajanju sastojaka uzoraka nerastvorljivih u rastvoru hlorovodonične kiseline i na daljoj primeni postupka A.
Reagensi
Koriste se sledeći reagensi:
1) 10 %-ni rastvor hlorovodonične kiseline;
2) rastvor trihlorsirćetne kiseline (odmeri se 200 g kiseline i dopuni vodom do 1 l) - (za postupak A);
3) rastvor trihlorsirćetne kiseline (odmeri se 10 g kiseline i dopuni vodom do 10 l).
Aparati i pribor
Koriste se sledeći aparati i pribor:
1) mufolna peć, sa mogućnošću regulacije temperature na 550°C (temperatura u peći u blizini posuda za spaljivanje ne sme se razlikovati više od 20°C u odnosu na određenu temperaturu);
2) analitička vaga;
3) sušnica, sa mogućnošću regulacije temperature na 103 ± 2°C;
4) rešo ili plamenik;
5) vodeno kupatilo;
6) posude za sagorevanje od porculana, platine ili legure platine i srebra ili drugog pogodnog materijala, zapremine od 20 do 30 ml i visine do 5 cm;
7) eksikator sa efikasnim sredstvom za sušenje.
Određivanje
Postupak A
U ižarenu posudu odmeri se 5 g uzorka za ispitivanje, sa tačnošću od 0,0001 g. Posuda za sagorevanje, sa uzorkom za ispitivanje, stavi se na rešo ili plamenik i postepeno zagreva do ugljenisanja, a zatim se prenese u mufolnu peć prethodno zagrejanu na 550°C i ostavi 3 h. Vizuelno se proveri da li je pepeo oslobođen ugljenisanih čestica. Ako nije, posuda se ponovo vraća u peć i žari 1 h. Ako su ugljenisane čestice još vidljive ili ako postoji sumnja da ih još ima, pepeo se ohladi, a zatim se navlaži destilovanom vodom. Vlaga postepeno isparava u sušnici, na temperaturi 103 ± 2°C, pepeo se ponovo premesti u mufolnu peć i žari 1 h. Posuda se zatim stavi u eksikator da se ohladi do sobne temperature. Ostatak u posudi - pepeo kvantitativno se prenese u čašu zapremine od 250 do 400 ml, prelije sa 75 ml rastvora hlorovodonične kiseline, pažljivo greje na rešou ili plameniku do ključanja i ostavi da ključa 15 min. Vruć rastvor se filtrira preko filtrir-papira, bez pepela, zatim se filtrir-papir sa ostatkom ispira vrućom vodom sve dok se iz vode za ispitivanje ne odstrani kiselina. Filtrir-papir sa ostatkom prenese se u posudu za sagorevanje koja je prethodno 30 min zagrevana u mufolnoj peći na temperaturi 550°C, ohlađenu u eksikatoru i izmerenu sa tačnošću 0,001 g. Sadržaj u posudi se 2 h greje u sušnici na temperaturi 103 ± 2°C, zatim se 30 min žari u mufolnoj peći, na temperaturi 550°C. Posle žarenja posuda se stavlja u eksikator da se ohladi do sobne temperature i ohlađena se brzo izmeri, sa tačnošću 0,0001 g.
Postupak B
U posudu sa uzorkom za određivanje postepeno se dodaje 25 ml vode i 25 ml rastvora hlorovodonične kiseline, promeša i sačeka da se pena izgubi. Zatim se doda još 50 ml rastvora hlorovodonične kiseline, ponovo promeša i, po potrebi, sačeka da se pena izgubi. Posuda se 30 min ili duže greje na ključalom vodenom kupatilu dok se skrob ne razgradi.
Vruć rastvor se filtrira preko filtrir-papira, bez pepela, i filtrir-papir sa ostatkom ispira se sa 50 ml vruće vode.
Napomena. |
Ako se rastvor teško filtrira, određivanje se mora ponoviti na novom uzorku, ali se sada umesto 50 ml rastvora hlorovodonične kiseline doda 50 ml trihlorsirćetne kiseline, a i filtrir-papir sa ostatkom pepela, pre ispiranja toplom vodom, ispira se toplim rastvorom trihlorsirćetne kiseline. Filtrir-papir sa ostatkom pepela prenese se u posudu za žarenje, suši u sušnici 2 h na temperaturi 103 ± 2°C i zatim žari 3 h u mufolnoj peći. Posle žarenja posuda se stavi u eksikator da se ohladi do sobne temperature i ohlađena brzo meri, sa tačnošću od 0,0001 g. |
Izračunavanje
Količina pepela nerastvorljivog u hlorovodoničnoj kiselini izražava se u procentima mase i izračunava po sledećoj formuli:
(m2 - m0) · |
100 |
|
m1 |
gde je:
m0 - masa prazne posude, u gramima;
m1 - masa uzorka za ispitivanje, u gramima;
m2 - masa posude sa pepelom nerastvorljivim u hlorovodoničnoj kiselini, u gramima.
Kao rezultat uzima se srednja aritmetička vrednost dva paralelna određivanja, ako su ispunjeni zahtevi u pogledu ponovljivosti, i rezultat se izražava dvema decimalama (m/m).
Ponovljivost
Razlika između dva paralelna određivanja na istom uzorku ne sme biti veća od:
0,3 jedinice (u apsolutnoj vrednosti) - za sadržaj pepela nerastvorljivog u hlorovodoničnoj kiselini manji od 3 %;
0,4 jedinice (u apsolutnoj vrednosti) - za sadržaj pepela nerastvorljivog u hlorovodoničnoj kiselini od 3 do 5%;
0,5 jedinica (u apsolutnoj vrednosti) - za sadržaj pepela nerastvorljivog u hlorovodoničnoj kiselini od 5 do 20%;
0,7 jedinica (u apsolutnoj vrednosti) - za sadržaj pepela nerastvorljivog u hlorovodoničnoj kiselini od 20 do 40%;
1,0 jedinice (u apsolutnoj vrednosti) - za sadržaj pepela nerastvorljivog u hlorovodoničnoj kiselini veći od 40%.
20. Određivanje bezazotnih ekstraktivnih materija
Primena
Određivanje bezazotnih ekstraktivnih materija primenjuje se za sve vrste stočne hrane.
Izračunavanje
Bezazotne ekstraktivne materije izražavaju se u procentima mase uzorka i izračunavaju se po sledećoj formuli:
100 - (V + SP + SM + SC + MM)
gde je:
SP - procent sirovih proteina;
SM - procent sirovih masti;
SC - procent sirove celuloze;
V - procent vlage;
MM - procent mineralnih materija.
Princip i primena
Princip se sastoji u titraciji vodenog rastvora pripremljene količine uzorka rastvorom srebro-nitrata, koji sa hloridom daje u vodi talog srebro-hlorida.
Metoda se primenjuje za određivanje hlorida u stočnoj hrani.
Reagensi
Za određivanje bezazotnih ekstraktivnih materija koriste se sledeći reagensi, čistoće pro analysi, i destilovana voda:
1) 10 %-ni rastvor kalijum-hromata;
2) rastvor srebro-nitrata c(AgNO3) = mol/l.
Pribor
Pored uobičajene laboratorijske opreme, koristi se i sledeći pribor:
1) sud za odmeravanje;
2) stakleni sud, zapremine 100 ml;
3) pipeta, zapremine 20 ml;
4) staklena čaša, zapremine 400 ml;
5) špric-boca;
6) bireta;
7) stakleni štapić.
Određivanje
Na analitičkoj vagi odmeri se približno 10 g uzorka, sa tačnošću 0,001 g i doda 50 ml destilovane vode, dobro se promeša i ostavi 2 h uz povremeno mešanje. Zatim se procedi kroz filtrir-papir u stakleni sud zapremine 100 ml, talog se dobro ispere destilovanom vodom i posuda se do oznake dopuni destilovanom vodom. Od rastvora pripremljenog na taj način pipetom se uzme 20 ml, doda 2 do 5 kapi 10 %-nog kalijum-hromata i titrira sa 0,1 mol/l rastvorom srebro-nitrata sve dok rastvor ne postane oranž.
Izračunavanje
Količina natrijum-hlorida dobija se iz količine utrošenog rastvora srebro-nitrata i količine uzorka za analizu.
Količina natrijum-hlorida izražava se u procentima, a izračunava se po sledećoj formuli:
A · F · 0,005844 · V · 100 |
||
a · m |
gde je:
A - zapremina 0,1 mol/l rastvora srebro-nitrata, utrošena za titraciju, u mililitrima;
F - faktor 0,1 mol/l rastvora srebro-nitrata;
0,005844 - masa natrijum-hlorida ekvivalentna 1 ml 0,1 mol/l rastvora srebro-nitrata;
V - zapremina rastvora u mernom sudu;
100 - preračun na procent;
a - zapremina rastvora upotrebljena za titraciju;
m - masa odmernog uzorka za analizu, u gramima.
22. Određivanje natrijum-hlorida
Primena
Metoda se primenjuje za utvrđivanje natrijum-hlorida u kuhinjskoj - jodiranoj soli.
Reagensi
Za određivanje natrijum-hlorida koriste se sledeći reagensi:
1) rastvor srebro-nitrata c(AgNO3) = 0,1 mol/l;
2) 10 %-ni rastvor kalcijum-hromata (K2CrO4).
Pripremanje rastvora
Odmeri se 17 g srebro-nitrata i rastvori u 1 l vode, pa se titar ovog rastvora odredi pomoću rastvora hemijski čistog, osušenog NaCl, na temperaturi 105°C, na sledeći način: 0,1 do 0,2 g NaCl, izmerenog sa tačnošću 0,0002 g, prebaci se u koničnu tikvicu, rastvori se u 30 ml destilovane vode, doda se 4 kapi 10 %-nog rastvora kalijum-hromata i titrira pripremljenim rastvorom srebro-nitrata sve dok crvena boja ne bude postojana.
Određivanje
Odmeri se 5 g uzorka, prenese u mernu bocu zapremine 250 ml, prelije vodom do dve trećine zapremine boce, pa se povremeno, u toku pola sata, snažno mućka. Tada se dolije voda do oznake i filtrira kroz suvi nabran filtrir-papir. Prvi deo filtrata se odbaci, pa se zatim pipetom uzme 50 ml filtrata, koji se titrira rastvorom 0,1 mol (AgNO3)/l, uz dodavanje 2 do 3 kapi 10 %-nog rastvora kalijum-hromata sve dok crvenkasta boja ne bude postojana.
Izračunavanje
Sadržaj natrijum-hlorida izražava se u procentima i izračunava po sledećoj formuli:
a · f · 0,005846 · V · 100 |
||
b · m |
gde je:
a - količina rastvora 0,1 mol (AgNO3)/l, u mililitrima, utrošenog za titraciju;
f - faktor rastvora 0,1 mol (AgNO3)/l;
0,005846 - količina NaCl, u gramima, koja odgovara 1 ml tačno 0,1 mol/l rastvora AgNO3;
V - zapremina tečnosti u mernoj boci, u mililitrima;
b - količina filtrata uzeta za titraciju, u mililitrima;
m - masa ispitivanog uzorka, u gramima.
Napomena. |
Ako je filtrat koji se dobija u postupku određivanja tamno obojen, onda se alikvotni deo filtrata, uz dodavanje 1 ml 10 %-nog rastvora natrijum-karbonata, upari do suvog i žari u električnoj peći na temperaturi do 500°C .Beli do sivo beli pepeo rastvori se u razblaženoj azotnoj kiselini (1 + 3) i filtrira, a zatim se u filtratu odredi NaCl kao što je utvrđeno u postupku određivanja. |
23. Određivanje organoleptičkih svojstava jodirane soli za stočnu hranu
Određivanje mirisa
Odmeri se 20 g jodirane soli i sa malo vode rastrlja tučkom u tarioniku. Mešavina ne sme ispuštati miris, osim blagog specifičnog mirisa na materije ako su dodate radi oplemenjivanja soli ili u terapeutske svrhe.
Ispitivanje ukusa
Ukus se utvrđuje probom 5 %-nog vodenog rastvora soli u destilovanoj vodi.
Ispitivanje boje
Jodirana so mora imati ciglacrvenu, odnosno sivocrnu boju, koja mora odgovarati standardnim uzorcima soli denaturisane sa 0,2 % hematita u prahu ili fino isitnjenog drvenog uglja.
24. Određivanje vode u jodiranoj soli (higroskopnoj i kristalnoj)
Određivanje
U prethodno osušenu i tariranu staklenu posudu za merenje, sa brušenim poklopcem, izmeri se oko 5 g uzorka, tako da bude razastrt po celom dnu posude. Zajedno sa brušenim poklopcem posuda se unese u sušnicu i suši na temperaturi 105°C do konstantne mase (sušenje traje oko 3 h). Posle hlađenja od 30 min u eksikatoru sa kalcijum-hloridom ili silikagelu, posuda sa poklopcem se izmeri.
Izračunavanje
Količina vode izražava se u procentima mase i izračunava se prema sledećoj formuli:
b · 100 |
||
a |
gde je:
b - gubitak u masi soli posle sušenja, u gramima;
a - izmerena količina uzorka, u gramima.
25. Određivanje materija nerastvorljivih i rastvorljivih u vodi u jodiranoj soli
Pripremanje uzorka
Za određivanje materija nerastvorljivih i rastvorljivih u vodi, uzorak se priprema tako što se uzorak za ispitivanje isitni da bi mogao da prođe kroz sito sa otvorima od 1 mm.
ODREĐIVANJE MATERIJA NERASTVORLJIVIH U VODI
Odmeri se 10 g jodirane soli i rastvori u 200 ml destilovane vode. Zatim se zagreje do vrenja i odmah filtrira kroz gust filtrir-papir, koji je prethodno ispran 96 %-nim etanolom a zatim se 20 min suši na temperaturi 105°C i izmeri.
Ostatak na filtru ispere se vrućom vodom do prestanka reakcije na hlor. Posle toga se filtrir-papir sa nerastvorljivim ostatkom suši 2 h na temperaturi 105°C, ohladi u eksikatoru i meri. Filtrat se hvata u mernu bocu zapremine 500 ml. Kad se ohladi, dopuni se do oznake destilovanom vodom i čuva za dalje ispitivanje.
Količina materija nerastvorljivih u vodi izražava se u procentima i izračunava se po sledećoj formuli:
100 · b |
||
a |
gde je:
a - masa uzorka za analizu, u gramima;
b - masa ostatka na filtrir-papiru, u gramima.
ODREĐIVANJE MATERIJA RASTVORLJIVIH U VODI
Iz filtrata dobijenog odvajanjem sastojaka nerastvorljivih u vodi određuju se sledeći sastojci jodirane soli: SO42-, Cl, Ca, Mg i MgCl2.
ODREĐIVANJE JONA SO42- - GRAVIMETRIJSKA METODA
Princip
Princip se zasniva na taloženju sulfatnih jona u rastvoru hlorovodonične kiseline kao barijum-sulfata, koji se i žari i meri.
Reagensi
Za određivanje jona SO42- koriste se sledeći reagensi:
- 38 %-na hlorovodonična kiselina (r = 1,19 g/ml);
- rastvor barijum-hlorida: odmeri se 122 g BaCl2 · 2H2O i rastvori u 1 000 ml vode.
Određivanje
Rastvori se 5 g uzorka za ispitivanje u 100 ml destilovane vode, kojoj je dodato nekoliko mililitara hlorovodonične kiseline. Sve se zagreje do ključanja i doda topli rastvor barijum-hlorida u malom višku, pa se ostavi nekoliko sati da se staloži. Tada se profiltrira kroz gust filtrir-papir, ispere vrućom destilovanom vodom dok ne iščezne reakcija na Cl-, osuši u lončiću, žari na temperaturi 600°C, ohladi u eksikatoru i meri:
procent SO42- = 8,231 · a;
procent SO3 = 6,86 · a;
gde je:
a - sadržaj lončića, u gramima.
ODREĐIVANJE JONA HLORA
Posle određivanja materija nerastvorljivih u vodi, iz svake merne boce otpipetira se po 10 ml filtrata u konične tikvice zapremine 300 ml i dopuni destilovanom vodom do 100 ml, doda 6-7 kapi 5%-nog rastvora K2CrO4 i titrira se 0,1 mol (AgNO3)/l do prve najmanje promene boje.
Količina hlora izražava se u procentima i izračunava po sledećoj formuli:
b · 50 · 0,35457 |
||
a |
gde je:
a - masa izmerene soli, u gramima;
b - utrošak 0,1 mol (AgNO3)/l, u mililitrima.
IZRAČUNAVANJE NATRIJUM-HLORIDA
Količina NaCl izražava se u procentima i izračunava se po sledećoj formuli:
5,845 · a
gde je:
a - broj utrošenih mililitara rastvora AgNO3 za titraciju.
ODREĐIVANJE MEGNEZIJUM-HLORIDA
Princip
Metoda se zasniva na ekstrakciji uzorka za ispitivanje etanolom i titraciji dinatrijum-etilen-diamin-tetrasirćetnom kiselinom (kompleksom III).
Reagensi
Za određivanje magnezijum-hlorida koriste se sledeći reagensi:
1) etanol 96 %-ni;
2) rastvori za titraciju;
a) rastvor natrijum-hidroksida c(NaOH) = 1 mol/l;
b) 0,1 %-ni vodeni rastvor metil-oranža;
v) rastvor hlorovodonične kiseline c(HCl) = 0,1 mol/l;
g) rastvor dinatrijum-etilen-diamin-tetrasirćetne kiseline c = 0,05 mol/l (EDTA);
d) puferni rastvor pH = 10 (54 g NH4Cl + 350 ml 25%-nog rastvora NH4OH dopuni se destilovanom vodom do 1 000 ml);
đ) eriohrom-crno T: fino se isitni 1 g eriohrom-crnog sa 100 g NaCl.
Određivanje
Odmeri se 10 g uzorka, stavi u bocu zapremine 200 ml, doda se 50 ml etanola, zatvori i mućka 10 min, a zatim se filtrira. Pipetom se prenese 25 ml filtrata u koničnu tikvicu, doda se 70 ml destilovane vode, 5 ml pufernog rastvora i oko 0,3 g smeše indikatora i titrira rastvorom kompleksona III do promene boje u plavo.
Izračunavanje
Količina MgCl2 izražava se u procentima i izračunava po sledećoj formuli:
0,09253 · a
gde je:
a - broj mililitara rastvora utrošenog pri titraciji kompleksonom III.
ODREĐIVANJE KALCIJUMA
Posle određivanja materija nerastvorljivih u vodi, od filtrata se otpipetira 50 ml u koničnu tikvicu zapremine 300 ml. Posle dodavanja 10 - 15 ml 0,1 mol (NaOH)/l i indikatora (mureksid i NaCl u odnosu 1.250), titrira se sa 0,01 mol/l kompleksona III do promene boje iz ružičaste u ljubičastu.
Količina Ca izražava se u procentima i izračunava po sledećoj formuli:
Ca = |
0,04 · b · 100 |
|
a |
gde je:
a - količina ispitivane soli, u gramima,
b - broj mililitra rastvora utrošenog za titraciju.
ODREĐIVANJE MAGNEZIJUMA
Posle određivanje materija nerastvorljivih u vodi, od filtrata se otpipetira 50 ml u koničnu tikvicu zapremine 300 ml i zagreje do temperature 40°C, doda se 5 ml pufernog rastvora i indikator eriohrom-crno T. Posle toga se rastvor, čija je pH vrednost 10, titrira rastvorom 0,01 mol (komplekson III/1) do promene boje u svetlo-plavu.
Količina Mg izražava se u procentima i izračunava po sledećoj formuli:
(c-b)1,22 |
||
a |
gde je:
a - količina ispitivane soli, u gramima;
b - utrošak rastvora za titraciju Ca, u mililitrima;
c - utrošak rastvora za titraciju Ca + Mg, u mililitrima.
26. Određivanje natrijuma i kalijuma
Princip i primena
Princip se zasniva na određivanju natrijuma i kalijuma pomoću plamenog fotometra. Metoda za određivanje kalijuma i natrijuma primenjuje se za stočna hraniva, hranljive dodatke i krmne smeše.
Pripremanje rastvora uzorka za fotometrisanje
Odmeri se oko 1 g uzorka, navlaži sumpornom kiselinom (razblaženom 1 + 10), osuši na električnoj ploči i žari na temperaturi od približno 500°C do potpune razgradnje organske materije. Ostatak se rastvori kuvanjem sa 2 do 5 ml hlorovodonične kiseline na vodenom kupatilu, uz dodavanje 50 ml vode. Sadržaj posude prebaci se u čašu, zagreje do ključanja i amonijak dodaje do potpunog taloženja gvožđa, aluminijuma i fosfata. U rastvoru mora biti amonijak u neznatnom višku (miris). Neposredno posle toga vrši se filtriranje, pa se talog ispere vrućom vodom. Talog se, kad ga ima u većim količinima, rastvori u što manje hlorovodonične kiseline, ponovo taloži sa amonijakom i dobijeni talog ispira vrućom vodom. Sjedinjeni filtrati (ako je potrebno) upare se na manju zapreminu, prespu u mernu bocu i dopune vodom do oznake. U rastvoru se nalaze ukupne količine kalijuma i natrijuma.
Određivanje
ODREĐIVANJE KALIJUMA
Emisioni intenzitet kalijumovog spektra određuje se monohromatorom na 768 nm (nanometara) ili upotrebom nekih standardnih filtara, u plamenu acetilena i vazduha ili kiseonika, ili vodonika i kiseonika. Rastvor uzorka pripremljen na taj način stavi se u raspršivač instrumenta, pa se na galvanometru odredi skretanje kazaljke koje treba da se nađe u granicima određenim baždarnom krivom. U protivnom, potrebno je da se rastvor razblaži. Baždarna kriva određuje se na taj način što se 1,5829 g kalijum-hlorida (sušenog na temperaturi 500°C) rastvori u 1 000 ml vode (1 ml rastvora sadrži 1 mg K2O), pa se od tog rastvora uzme, prema očekivanom sadržaju kalijuma u uzorku: 10, 20 ml itd. i razblaži vodom do 1 000 ml. Tako dobijenim rastvorima odredi se plamen-fotometrom intenzitet skretanja galvanometra za talasnu dužinu od 768 nm. Dobijene vrednosti za baždarnu krivu (propustljivost) nanesu se na milimetarski papir, i to: na ordinatu-fotometrijske vrednosti, a na apscisu-odgovarajuće koncentracije standardnih rastvora kao K2O, u µg/ml. Kroz tako dobijene tačke provuče se baždarna kriva. Iz veličine fotometrijske vrednosti uzorka, a pomoću baždarne krive, odredi se sadržaj K2O u uzorku K = 0,83013 K2O.
ODREĐIVANJE NATRIJUMA
Emisioni intenzitet natrijumovog spektra određuje se monohromatorom na talasnoj dužini 589,3 nm ili korišćenjem nekih standardnih filtara: u plamenu acetilena i vazduha ili kiseonika, gasa ili vazduha ili kiseonika, ili vodonika i kiseonika. Rastvor uzorka, pripremljen kao za određivanje kalijuma, stavi se u raspršivač, pa se na galvanometru instrumenta odredi skretanje kazaljke, koje treba da bude u granicama određenim baždarnom krivom.
Baždarna kriva se određuje na taj način što se 1,8858 g natrijum-hlorida, koji je sušen na 180°C, rastvori u 1000 ml vode (1 ml rastvora sadrži 1 mg N2O), pa se od tog rastvora uzme, prema očekivanom sadržaju natrijuma u uzorku: 10, 20, 30 ml itd. i razblaži vodom do 1 000 ml. U tako dobijenim rastvorima odredi se plamen-fotometrom intezitet skretanja za talasnu dužinu od 589,3 nm. Dobijene vrednosti za baždarnu krivu nanesu se na milimetarski papir, i to: na ordinatu - fotometrijske vrednosti, a na apscisu - odgovarajuće koncentracije standardnih rastvora kao N2O u µg/ml. Kroz tako dobijene tačke povuče se baždarna kriva. Iz veličine fotometrijske vrednosti uzorka, pomoću baždarne krive odredi se sadržaj N2O u uzorku: Na = 0,74191 N2O.
27. Određivanje kalcijuma - kompleksometrijska metoda
Princip i primena
Metoda se može primenjivati za određivanje kalcijuma u stočnoj hrani: stočnim smešama, kabastoj hrani, koncentratima i sirovinama za smeše.
Posle spaljivanja uzorka, dobijeni pepeo se rastvori i alikvotni deo tretira se dinatrijumovom soli etilen-diamin-tetrasirćetne kiseline (EDTA), uz prisustvo smeše indikatora (kalceina i timolftaleina). Da bi se postigla bolja indikacija ekvivalentne tačke, u prvom se stepenu dodaje višak EDTA, koji se retitrira rastvorom kalcijuma.
Reagensi
Koriste se sledeći reagensi, čistoće pro analysi:
1) rastvor natrijum-hidroksida c(NAOH) = 1 mol/l (40 g NaOH rastvori se u 1 000 ml vode);
2) rastvor dinatrijumove soli etilen-diamin-tetrasirćetne kiseline;
a) rastvor dinatrijumove soli etilen-diamin-tetrasirćetne kiseline c(EDTA) = 0,1 mol/l (rastvori se 37,221 g EDTA u vodi, kvantitativno se prenese u odmernu tikvicu zapremine 1 000 ml i dopuni vodom do oznake). Taj rastvor se najbolje čuva u polietilenskoj boci, i to na tamnom mestu;
b) rastvor dinatrijumove soli etilen-damin-tetrasirćetne kiseline c(EDTA) = 0,01 mol/l (200 ml 0,1 mol/l rastvora EDTA razblaži se sa 1 800 ml čiste vode, dobro izmeša i upotrebi za titraciju. Koncentracija rastvora se svakodnevno kontroliše prilikom upotrebe);
3) rastvor kalcijuma za retitraciju;
a) primenjuje se standardni rastvor za određivanje tvrdoće vode, titrival (1 ml = 1 mg CaO po razblaženju na 1 I) i razblaži se destilovanom vodom do 2 000 ml, pri čemu se dobija rastvor 0,008915 mol/l, ili
b) odmeri se tačno 17,846 g suvog kalcijum-karbonata, kvantitativno prenese sa destilovanom vodom u od-mernu tikvicu zapremine 1 l, pažljivo se dodaje toliko 25%-ne hlorovodonične kiseline koliko je potrebno da prestane izlučivanje ugljen-dioksida, a zatim istom vodom dopuni do oznake. Rastvor se čuva u dobro zatvorenoj staklenoj boci i služi za pripremanje radnog rastvora. Odmeri se tačno 50 ml tog rastvora, prenese u odmernu bocu zapremine 1 I i dopuni vodom do oznake. Koncentracija tako razblaženog rastvora iznosi 0,008915 mol/l;
4) indikatorska mešavina, odmeri se 0,2 g kalceina, 0,12 g timolftaleina i 20 g kalijum-nitrata pa se u tarioniku dobro izmeša i isitni. Čuva se u dobro zatvorenoj boci na suvom mestu (najbolje u eksikatoru);
5) kalijum-nitrat;
6) 25%-ni rastvor hlorovodonične kiseline.
Aparatura i pribor
Osim uobičajene laboratorijske opreme, koristi se i:
1) staklena čaša, zapremine 250 ml;
2) automatska staklena bireta, zapremine 25 ml i 50 ml;
3) magnetna mešalica;
4) stona lampa, jačine 60 - 100 W;
5) pipeta trbušasta, zapremine 1 ml; 2 ml; 10 ml; 20 ml i 50 ml;
6) filtrir-papir (bela traka).
Određivanje
Odmerena količina uzorka žari se na temperaturi od 540 do 600°C. Uzorke koji ne sadrže organske materije nije potrebno žariti, već samo odmeriti i rastvoriti u kiselini. Pripremljeni pepeo od stočne hrane navlaži se vodom i u lončić zapremine 5 ml dodaje se 25%-ne hlorovodonične kiseline, a zatim se tretira na vrelom vodenom kupatilu da hlorovodonična kiselina ispari. Ponovo se doda 2 ml hlorovodonične kiseline, a nekoliko minuta kasnije i destilovana voda (malo više od polovine) i ostavi se na kupatilu samo toliko da se rastvor zagreje, a da ne ispari.
Rastvor se zatim, preko levka s filtrir-papirom, kvantitativno prenese u odmernu tikvicu zapremine 100 ml i do oznake dopuni destilovanom vodom.
Alikvotan deo rastvora pepela (a), koji sadrži 2 do 8 mg Ca, odmeri se trbušastom pipetom u čašu za titraciju na magnetnoj mešalici. Zatim se doda oko 50 mg indikatorske mešavine, 5 ml 1 mol/l rastvora natrijum-hidroksida i destilovane vode do približno 80 ml. Posle toga rastvor treba da bude ljubičaste boje, sa tamnozelenom fluorescencijom. Ako je rastvor žutozelene boje, ponovo se dodaje 5 ml natrijum-hidroksida.
Uz intenzivno mešanje magnetnom mešalicom dodaje se iz birete 0,01 mol/l rastvora EDTA sve dok se ne izgubi zelena fluorescencija i dok se ne intenzivira boja osnovnog rastvora. Dozira se višak EDTA od najmanje 5 ml, sačeka najmanje pola minuta, a zatim se višak EDTA retitrira sa 0,008915 mol/l rastvora kalcijuma, sve dok se ne pojavi zelena fluorescencija (osnovna ljubičasta boja izbledi). Ako se rastvor pretitrira, ponovo se dodaje EDTA, pa se ekvivalentna tačka može veoma tačno odrediti. Na kraju se zapiše količina utrošenog EDTA (V1) i rastvora kalcijuma (V2) u mililitrima.
Koncentracija EDTA kontroliše se tako što se u čašu odmeri 60 ml destilovane vode, indikator i 5 ml NaOH. Iz birete se dozira oko 15 ml EDTA, pa se zatim titrira rastvorom kalcijuma do pojave zelene fluorescencije. Količina utrošenog EDTA (V1) i rastvora kalcijuma (V2), u mililitrima, zapiše se pa se izračunava potrošnja EDTA na 1 ml rastvora kalcijuma. Taj faktor određuje se u tri ponavljanja, sa tačnošću na četvrtoj decimali.
Izračunavanje
Količina kalcijuma izražava se u procentima i izračunava se po sledećoj formuli:
(V1 - V2 · f) · |
3,5731 |
· a · m |
f |
gde je:
V1 - utrošen rastvor 0,01 mol/l EDTA za titraciju uzorka, u mililitrima;
V2 - utrošen rastvor 0,008915 mol/l rastvora Ca za retitraciju, u mililitrima;
f = V1/V2 - faktor koji predstavlja potrošnju EDTA u mililitrima na 1 ml rastvora kalcijuma pri određivanju koncentracije EDTA;
a - odmereni broj mililitara rastvora uzorka uzetog za titraciju;
m - masa uzorka za pripremanje pepela, u gramima.
Ponovljivost
Određivanje se vrši u dve paralelne probe. Razlika između rezultata dva paralelena određivanja ili ubrzo jednog za drugim iznosi 3% a za kabastu stočnu hranu - najviše 5%.
Napomena. |
Ako se za utvrđivanje pepela koristi 3 g uzorka, pri određivanju kalcijuma za pojedine vrste stočne hrane uzima se sledeći alikvot (a) za titraciju: |
|
|
kukuruz i silaža |
30 do 50 ml; |
|
seno, trava |
20 do 30 ml; |
|
smeša |
10ml; |
|
smeše za nosilje |
5 do 10 ml; |
|
mesno brašno |
5 do 20 ml; |
|
riblje brašno |
2 do 5 ml; |
|
koštano mesno brašno |
1 do 3 ml. |
|
Ekvivalentna tačka najbolje se uočava ako se titracija izvodi u tamnom delu laboratorije, na crnoj pozadini, uz konstantno direktno osvetljenje rastvora stonom lampom jačine 60 W do 100 W. |
|
Napomena. |
Prilikom titracije uzorka, rastvor EDTA dodaje se u višku od najmanje 5 ml, a zatim se do retitracije sačeka najmanje pola minuta da bi se obavila reakcija Ca u prisustvu fosfora. Ako se u to vreme ponovo javi zelena fluorescencija, doda se još najmanje 5 ml EDTA. |
|
Primena
Metoda za određivanje sadržaja megnezijuma primenjuje se za ispitivanje stočne hrane.
Određivanje
Izmeri se 1 do 5 g uzorka u platinski ili porculanski lončić i žari na temperaturi od približno 500°C, sve dok boja pepela ne postane bela do sivobela. Posle žarenja, pepeo se rastvori sa 5 do 10 ml hlorovodonične kiseline, prokuva se i filtrira. Talog se ispere, a filtrat potpuno ispari na vodenom kupatilu i suši u termostatu 3 h na temperaturi 120°C. Posle toga se doda 2 ml hlorovodonične kiseline i 50 ml vruće vode, prokuva se i filtrira. Talog se pere vrućom vodom. Filtrat se neutrališe amonijakom (1 + 1), pri čemu nastaje pahuljasti talog hidroksida gvožđa, aluminijuma, kalcijuma, magnezijuma i fosfata. Talog može izostati ako se pomenuti elementi nalaze u manjim količinama. Potrebna količina amonijaka određuje se po mirisu ili dodavanjem indikatora (metil-crveno).
U neutralisani rastvor doda se 1 ml (razblažene 1 + 1) sirćetne kiseline (kalcijum i magnezijum fosfat se rastvaraju) i 5 g natrijum-acetata ili amonijum-acetata. Rastvor se zagreje do temperature 80°C, pa se, ako je potrebno, dodaje kap po kap 10 %-nog rastvora ferihlorida (FeCl3). Potrebna količina ferihlorida određuje se promenom boje taloga koji nastaje dodavanjem ferihlorida u rastvor. Čim se počne stvarati crvenosmeđi talog, treba prestati sa dodavanjem ferihlorida i sva količina fosfata se odstrani.
Posle nekoliko minuta talog se odstrani filtriranjem preko filtrir-papira, pa se talog pere vrućim rastvorom amonijum-acetata. U bistar filtrat doda se 10 ml zasićenog rastvora amonijum-oksalata [(NH4)2 C2O4] i 1 do 2 kapi indikatora (metil-crveno). Filtrat se zagreje skoro do ključanja i postepeno neutrališe amonijakom, dok crvena boja indikatora ne pređe u žutu boju. Posle toga ostavi se 4 h na vrućem vodenom kupatilu, a zatim se izdvojeni talog filtrira. Talog i filtrat se isperu vrućom vodom.
Filtrat i voda, posle pranja taloga, potpuno se upare uz dodavanje 3 ml azotne kiseline, pa se zatim amonijačne pare odstrane opreznim zagrevanjem. Ostatak se rastvori u 5 ml hlorovodonične kiseline i doda destilovana voda do približno 100 ml. Posle toga doda se 5 ml 10 %-nog rastvora natrijum-citrata i 10 ml 10 %-nog rastvora amonijum-fosfata [(NH4)2 HPO4] ili toliko da se sav magnezijum istaloži. Zatim se doliva amonijak (razblažen 1 + 4), uz stalno mešanje staklenim štapićem, dok rastvor ne postane alkalan i dok se ne počne stvarati talog amonijum-fosfata. Tada se naglo doda 25 ml amonijaka (1 + 4) i meša do potpunog taloženja magnezijuma. Talog se ostavi preko noći na hladnom, pa se zatim filtrira preko filtrir-papira. Dobijeni talog se ispere hladnim amonijakom (1 + 10) dok voda posle pranja ne počne da više reaguje na hloride. Talog se zatim žari na temperaturi od 900 do 1000°C, hladi u eksikatoru i meri kao magnezijum pirofosfat (Mg2P2O7).
Izračunavanje
Količina MgO izražava se u procentima i izračunava se po sledećoj formuli:
b · 0,36226 |
· 100 |
|
m |
gde je:
b - masa dobijenog taloga, u gramima;
m - masa ispitivanog uzorka, u gramima;
0,36226 - faktor za preračunavanje MgO iz Mg2O2O2O7.
29. Određivanje ukupnog fosfora - spektrofotometrijska metoda
Princip i primena
Metoda se primenjuje pri utvrđivanju ukupnog sadržaja fosfora za sve vrste stočne hrane.
Princip se zasniva na spaljivanju uzorka (kad je hrana organskog porekla) ili vlažnom spaljivanju (kad je hrana mineralnog porekla), zatim na rastvaranju u kiselom rastvoru. Rastvor se tretira molibden-vanadatnim reagensom i apsorbancija dobijenog žutog rastvora meri se spektrofotometrom na talasnoj dužini od 430 nm.
Reagensi
Za određivanje sadržaja ukupnog fosfora potrebni su sledeći reagensi, čistoće pro analysi, i destilovana voda:
1) kalcijum-karbonat;
2) hlorovodonična kiselina c(HCl) = 6 mol/l;
3) azotna kiselina c(HNO3) = 1 mol/l;
4) azotna kiselina, r20 = 1,38 g/ml;
5) sumporna kiselina, r20 = 1,84 g/ml;
6) molibden-vanadatni reagens: u odmernu tikvicu zapremine 1 l promeša se 200 ml rastvora amonijum-heptomolibdata, 200 ml rastvora amonijum-monovanadata i 135 ml azotne kiseline (r20 = 1,38 g/ml) i do oznake dopuni destilovanom vodom;
7) rastvor amonijum-heptomolibdata: u vrućoj destilovanoj vodi rastvori se 100 g heptomolibdat-tetrahidrata (NH4)6 Mo7 O24 · 4H2O), doda 10 ml amonijum-hidroksida (r20 = 0,91 g/ml) i do 1 l dopuni destilovanom vodom;
8) rastvor amonijum-monovanadata: u 400 ml vruće vode rastvori se 2,35 g amonijum-monovanadata (NH4VO3), zatim se uz stalno mešanje postepeno dodaje 20 ml razblažene azotne kiseline [7 ml azotne kiseline (r20 = 1,38 g/ml) + 13 ml destilovane vode] i dopuni destilovanom vodom do 1 I;
9) standardni rastvor fosfora koji sadrži 1 mg fosfora/ml: u odmernoj tikvici zapremine 1 l rastvori se u destilovanoj vodi 4,367 g kalijum-dihidrogen-fosfata (KH2PO4) i do oznake dopuni destilovanom vodom.
Aparatura i pribor
Pored uobičajene laboratorijske opreme, potreban je i sledeći pribor:
1) lončić za žarenje od porculana ili kvarca;
2) električna mufolna peć sa regulacijom na 550 ± 20°C;
3) kjeldal-tikvica, zapremine 250 ml;
4) odmerne tikvice, zapremine 500 i 1000 ml;
5) pipete, graduisane;
6) spektrofotometar sa kivetama, zapremine 10 mm;
7) staklene cevi, kapaciteta od 25 do 30 ml, snabdevene staklenim zapušačima;
8) analitička vaga;
9) peščano kupatilo;
10) čaša, zapremine 250 ml.
Određivanje
Pripremanje rastvora za ispitivanje
Zavisno od prirode uzorka, rastvor za ispitivanje priprema se prema niže navedenim uslovima (suvo spaljivanje uzorka ili vlažno spaljivanje).
Suvo spaljivanje uzorka (za uzorke koji sadrže organsku materiju i od kojih se posle spaljivanja do suvog dobija nerastvorljivi ostatak)
U lončić za žarenje izmeri se oko 2,5 g uzorka, sa tačnošću od 0,001 g. Odmerena količina uzorka dobro se izmeša sa 1g kalcijum-karbonata i spaljuje u mufolnoj peći na temperaturi 550 ± 20°C sve dok se ne dobije beli ili sivi pepeo.
Pepeo se prenese u čašu zapremine 250 ml, doda se 20 ml vode i hlorovodonične kiseline do potpune neutralizacije. Zatim se doda još 10 ml 6 mol/l hlorovodonične kiseline, a čaša se stavi na peščano kupatilo i isparava do suvog ostatka da bi se dobio nerastvorljiv pesak. Zatim se ostavi da se hladi. Ostatku se doda 10 ml mol/1l azotne kiseline i stavi na peščano kupatilo da ključa 5 min. Tečnost se dekantuje u odmernu tikvicu zapremine 500 ml pri čemu se čaša više puta, ispira vrućom vodom, ostavi da se ohladi, dopuni vodom do oznake, promeša i filtrira.
Vlažno spaljivanje uzorka (za uzorke mineralnog porekla i uzorke u tečnom stanju)
Izmeri se 1 g uzorka za ispitivanje, sa tačnošću od 0,001 g, i prenese u kjeldal-tikvicu zapremine 250 ml, doda 20 ml sumporne kiseline i promućka tako da uzorak potpuno prekrije kiselina i da se spreči lepljenje za zidove tikvice. Tikvica sa sadržajem zagreva se do ključanja i sadržaj ključa 10 min. Zatim se ostavi da se postepeno hladi, doda se 2 ml azotne kiseline (r20 = 1,38 g/ml) i postepeno zagreva, ponovo postepeno hladi i doda malo azotne kiseline (r20 = 1,38 g/ml), pa se tako zagreva do ključanja.
Taj postupak se ponavlja sve dok se ne dobije obezbojen rastvor.
Rastvor se ohladi, doda malo destilovane vode i tečnost dekantacijom prenese u odmernu posudu zapremine 500 ml, pri čemu se kjeldal-tikvica ispira vrućom destilovanom vodom.
Rastvor se u odmernoj tikvici ohladi, zatim se do oznake dopuni destilovanom vodom, promućka i filtrira.
Razvijanje boje i merenje apsorbancije
Uzme se alikvotni deo filtrata dobijen postupkom pripremanja rastvora za ispitivanje da bi se dobila koncentracija fosfora najviše do 40 µg/ml.
10 ml ovog rastvora prenese se u cev za određivanje i doda 10 ml molibden-vanadatnog reagrensa, promućka i ostavi 10 min na sobnoj temperaturi. Zatim se na spektrofotometru meri apsorbancija obojenog rastvora na talasnoj dužini od 430 nm, u odnosu na slepu probu.
Napomena. Na istom uzorku vrše se dva određivanja.
Pripremanje standardne krive
Za pripremanje standardne krive koriste se standardni rastvori fosfora, koji se dalje pripremaju tako da sadrže 5; 10; 20; 30 i 40 µg fosfora/ml.
Od svakog rastvora uzme se 10 ml i svakome doda 10 ml molibden-vanadatnog reagensa, rastvori se promućkaju i ostave najmanje 10 min na sobnoj temperaturi, a zatim se apsorbancije mere spektrofotometrijski.
Nacrta se standardna kriva, pri čemu se vrednost apsorbancije nanosi na ordinatu, a na apscisu - odgovarajuće koncentracije fosfora µg/ml.
Slepa proba
Paralelno sa određivanjem priprema se i slepa proba, pri čemu se primenjuje isti postupak i koriste iste količine svih reagensa, ali bez uzorka.
Izračunavanje
Sadržaj ukupnog fosfora izražava se u procentima mase i izračunava po sledećoj formuli:
X · 500 · F · 100 |
= |
X · F |
||
m · 106 |
20 · m |
gde je:
X - sadržaj fosfora µg/ml, u razblaženom alikvotnom delu rastvora uzorka, pročitanom sa standardne krive;
m - masa uzorka koja je uzeta za pripremanje rastvora za ispitivanje, u gramima;
F - recipročan faktor razblaženja za alikvotan deo u postupku razvijanja boje i merenja apsorbancije.
Kao rezultat se uzima aritmetička sredina rezultata iz dva paralelna određivanja ako su zadovoljeni uslovi ponovljivosti.
Rezultat se ne sme razlikovati više od:
0,01 % (m/m) fosfora - za sadržaj fosfora manji od 3 % (m/m):
0,1 % (m/m) fosfora - za sadržaj fosfora jednak ili veći od 3% (m/m).
Ponovljivost
Razlika između rezultata dva paralelna određivanja koja je istovremeno ili neposredno jedno za drugim izveo isti analitičar ne sme biti veća od:
3 % (relativna vrednost) - za sadržaj fosfora jednak 5% ili veći od 5 % (m/m);
0,15 % (apsolutna vrednost) - za sadržaj fosfora jednak 5 % ili veći od 5% (m/m).
Princip i primena
Princip se zasniva na određivanju sumpora oksidacijom ispitivanog uzorka magnezijum-nitratom.
Metoda za određivanje sumpora primenjuje se pri ispitivanju stočnih hraniva, hranljivih dodataka i krmnih smeša.
Reagensi
Za određivanje sadržaja sumpora potrebni su sledeći reagensi:
1) rastvor magnezijum-nitrata: odmeri se 15 % MgO i doda 85 g HNO3, a zatim razblaži vodom u odnosu 1:1;
2) koncentrovana hlorovodonična kiselina;
3) 10%-ni rastvor barijum-hlorida.
Aparatura i pribor
Pored uobičajene laboratorijske opreme, koristi se i:
1) porculanski lončić;
2) vodeno kupatilo;
3) peć za žarenje;
4) staklena čaša, zapremine 250 ml;
5) merna boca, zapremine 250 ml.
Pripremanje uzorka
Odmeri se 1 g uzorka u porculanskom lončiću, doda se 7,5 ml rastvora magnezijum-nitrata i dobro promeša. Porculanski lončić sa uzorkom stavi se na vodeno kupatilo i dodaje kap po kap hlorovodonične kiseline dok se reakcija ne završi, pa se topli lončić prebaci u hladnu peć za žarenje i postepeno zagreva do temperature 500°C , do potpune oksidacije organske materije uzorka. Lončić se zatim izvadi iz peći i ostavi da se ohladi, a sadržaj se prebaci u čašu zapremine 250 ml i doda dovoljna količina vode i hlorovodonične kiseline u malom višku. Rastvor se zagreje do ključanja i filtrira, a talog se dobro ispere. Filtrat se prebaci u mernu bocu zapremine 250 ml, dopuni vodom do oznake, pa se u alikvotnom delu odredi sumpor kao što je navedeno u postupku određivanja.
Određivanje
U alikvotnom delu filtrata (10 do 200 ml) određuje se sumpor taloženjem. Alikvotni deo se otpipetira u čašu i doda vode do 200 ml, a zatim i toliko hlorovodonične kiseline da u rastvoru bude još oko 0,5 ml slobodne hlorovodonične kiseline. Zatim se zagreva do ključanja i doda 10 ml 10%-nog rastvora barijum-hlorida, uz konstantno mešanje. Kuvanje se nastavlja još 5 min, pa se ostavi 5 h ili duže da se taloži na vodenom kupatilu. Talog barijum-sulfata (BaSO4) filtrira se preko gustog filtrir-papira i ispira vrućom vodom sve dok posle ispiranja taloga voda ne prestane da pokazuje reakciju na hloride. Talog se zatim prebaci u žareni i izmereni porculanski lončić, suši se, žari se na što nižoj temperaturi, hladi se u eksikatoru i ponovo meri kao BaSO4.
Izračunavanje
Količina sumpora izražava se u procentima i izračunava po sledećoj formuli:
b 0,1374 |
·100 |
|
m |
gde je:
m - masa ispitivanog uzorka, u gramima;
b - masa BaSO4, u gramima.
Princip i primena
Za određivanje hlora primenjuju se dva postupka, i to: za određivanje ukupnog hlora - gravimetrijska metoda, a za određivanje rastvorljivog hlora - titrimetrijska metoda.
Postupci određivanja hlora primenjuju se pri određivanju hlora u stočnim hranivima, hranljivim dodacima i krmnim smešama.
ODREĐIVANJE UKUPNOG HLORA
Određivanje
Odmeri se 5 g uzorka za ispitivanje u platinskom lončiću, navlaži se sa 20 ml 5 %-nog rastvora natrijum-karbonata, upari do suvog i žari na što nižoj temperaturi, oko 500°C. Pepeo bele do sivobele boje ekstrahuje se vrućom vodom, filtrira i pere. Ako preostane veća količina nerastvorljivog pepela, onda se pepeo, zajedno sa filtrir-papirom, ponovo vrati u platinski lončić i žari na istoj temperaturi. Pepeo se rastvara u nekoliko mililitara azotne kiseline (1 + 4), filtracijom odvaja od nerastvorljivog dela, pere i doda vodenom ekstraktu.
U rastvor azotne kiseline doda se 10%-nog rastvora AgNO3 u malom višku. Rastvor se zagreje do ključanja, ostavi na tamnom mestu do potpune koagulacije taloga (AgCl) i filtrira preko lončića za filtriranje, a talog se pere vrućom vodom do uklanjanja viška AgNO3. Talog AgCl suši se na temperaturi od 140 do 150°C, hladi se i meri.
Izračunavanje
Količina ukupnog hlora izražava se u procentima i izračunava po sledećoj formuli:
0,24737 b |
· 100 |
|
m |
gde je:
m - masa ispitivanog uzorka, u gramima;
b - masa nađenog AgCl, u gramima.
ODREĐIVANJE RASTVORLJIVOG HLORA
Reagensi
Za određivanje sadržaja hlora potrebni su sledeći reagensi:
1) rastvor kalijum-hlorida: KCl suši se na temperaturi 500°C u peći za žarenje do konstantne mase, pa se 2,1026 g rastvori i razblaži vodom do 1000 ml; rastvor sadrži 0,001 g Cl/ml;
2) rastvor srebro-nitrata: odmeri se 5 g AgNO3 i rastvori u 1000 ml vode i podesi se tako da 1 ml tog rastvora bude ekvivalentan 1 ml standardnog rastvora KCl (titracijom);
3) rastvor kalijum-rodanida: odmeri se 2,5 g KCNS, rastvori u 1000 ml vode i podesi se tako da 1 ml tog rastvora odgovara 1 ml rastvora AgNO3; 40 do 50 ml rastvora srebro-nitrata titrira se uz indikator ferisulfat i 5 ml HNO3 (1 + 1) rastvorom rodanida, dok rastvor, posle jakog mućkanja, ne postane bledoružičast;
4) rastvor ferisulfata: odmeri se 6 g Fe2(SO4)3 · H2O i rastvori u destilovanoj vodi, pa se u mernoj boci doda vode do 100 ml;
5) rastvor indikatora ferisulfata: u 25 %-ni rastvor ferisulfata Fe2(SO4)3 · H2O doda se jednaka zapremina HNO3.
Određivanje
U koničnu tikvicu zapremine 300 ml odmeri se 2 do 5 g uzorka za ispitivanje i pipetom doda 50 ml rastvora Fe2(SO4)3. Za vreme dodavanja ferisulfata rastvor se lagano meša da bi se sprečilo grudvanje uzorka i olakšalo rastvaranje hlorida. Zatim se pipetom doda 100 ml amonijaka (razblaženog 1 + 19) i ponovo lagano meša, jer se rastvor vrlo teško filtrira ako se mućka. Rastvor se ostavi 10 min da se staloži, a zatim se filtrira preko filtrir-papira male gustoće. U 50 ml alikvotnog dela u uzorku sa 0 do 2% Cl ili u 25 ml u uzorku sa više od 2% hlora, hlor se određuje titracijom. Ako ni približna količina hlora u uzorku nije poznata, treba uzeti 10 ml filtrata radi eksperimentalne titracije. U taj filtrat dodaje se 10 ml HNO3 + 10 ml Fe2(SO4)3 (kao indikator) i razblaži do 50 ml. Posle toga doda se 0,5 ml rastvora kalijum-rodanida (KCNS) i neposredno, uz mešanje, toliko rastvora AgNO3 da se eliminiše crvena obojenost. Iz te približne titracije odredi se približna zapremina rastvora srebro-nitrata za potpuno taloženje hlora. U alikvotnom delu, koji će se uzeti za titraciju, potreban je višak od približno 10% rastvora azotne kiseline, mada i malo veći dodatak ne utiče na rezultat. Potrebno je uzeti najmanje 10 ml rastvora AgNO3.
U alikvotni deo, u čašu zapremine 250 ml, doda se 10 ml rastvora srebro-nitrata i 10 ml ferisulfata kao indikatora. Zatim se uz mešanje doda izračunata zapremina rastvora srebro-nitrata, zagreje do ključanja i ostavi da se ohladi do sobne temperature, uz mešanje, da bi talog koagulirao. Višak srebro-nitrata titrira se kalijum-rodanidom. Završna tačka je indicirana pojavom crvene boje, koja je postojana 15 sekundi.
Jedan mililitar utrošenog rastvora 0,1 mol/l srebro-nitrata odgovara 1 ml hlora.
Primena
Metoda za određivanje joda primenjuje se za ispitivanje joda u stočnim hranivima, hranljivim dodacima i krmnim smešama.
Aparati i pribor
Osim uobičajene laboratorijske opreme, koristi se i:
1) peć za spaljivanje (sl. 5 u prilogu): od gvozdenog lima načini se cilindar (1), dimenzija datih na slici, sa otvorom na vrhu koji je toliko širok da se u njega može staviti nikleni lončić (2) zapremine 100 ml. Ispod vrha cilindra montira se okrugla limena ploča (3) koja sprečava gasove plamena da direktno ližu zidove niklenog lončića. Pri dnu cilindra načini se dug uzan otvor (4) kroz koji se stavlja plamenik. Neznatno ispod gornje ivice cilindra načini se na plaštu osam otvora (5) za slobodno ispuštanje gasova koji se stvaraju pri sagorevanju.
Reagensi
Za određivanje sadržaja joda potrebni su sledeći reagensi:
1) 20 %-ni rastvor reducirane fosforne kiseline: nečistoće u fosfornoj kiselini, H3PO4, redukuju se razblaženjem 85 %-ne kiseline sa četiri zapremine vode i kratkotrajnim kuvanjem sa aluminijumskim listićima;
2) c(Na2S2O3) = 0,005 mol/l rastvor natrijum-tiosulfata: u čašu zapremine 25 ml otpipetira se 25 ml rastvora koji sadrži 0,1300 g KJ/1000 ml, doda se 200 ml H2O, 5 ml 20 %-nog rastvora NaHSO3 i 2 ili 3 g NaOH, neutrališe se sa H3PO4 i doda 1 ml u višku. Da bi se odredio broj miligrama joda koji je ekvivalentan sa dobijenim rastvorom Na2S2O3, primenjuje se sledeća formula:
f = |
2,5 |
|
ml 0,05 mol/l rastvora Na2S2O3 |
Rastvor natrijum-tiosulfata treba pripremiti istog dana kad se vrši određivanje joda u uzorku.
Određivanje
Odmeri se 20 g NaOH i 10 g KNO3 u niklenom lončiću, zajedno rastopi i zatim ohladi. Preko otopljene mase stavi se 1 do 10 g uzorka, koji se zatim navlaži sa 5 ml NaOH (1 + 1) i 10 ml 80 %-nog etanola. Lončić se stavi na hladnu električnu ploču, na kojoj, postepenim zagrevanjem, etanol ispari. Temeljno zagrevanje mase potrebno je da bi se izbeglo naknadno isparavanje i gubitak joda. Lončić se zatim stavi u spremljenu peć za spaljivanje i počne oprezno zagrevanje. Ako reakcija postane suviše burna, lončić se ukloni iz peći i neko vreme ostavi da se ohladi, a zatim se nastavi da se oprezno zagreva. Kad se otopljena masa potpuno umiri, naginjanjem lončića pokupe se sa strane i one čestice uzorka koje nisu regovale. U lončić se stavi nekoliko kristala KNO3 dok ne prestane dalje stvaranje gasova, a zatim se strane lončića isperu (naginjanjem lončića).
Lončić se na kraju izvuče iz peći, ohladi, stavi u čašu zapremine 600 ml i doda toliko vode da bude pokriven. Sadržaj lončića zagreva se kratko vreme, ali toliko da ne dođe do ključanja. Rastvor se ostavi preko noći, lončić se ispere i ukloni. Rastvor se neutrališe dodavanjem 10 ml koncentrovane H3PO4, ostavi 3 do 4 h na vodenom kupatilu, uz povremeno mešanje, da bi se jod potpuno rastvorio. Čaša se ohladi, nerastvoreni deo odstrani filtriranjem i talog ispere hladnom vodom. Zapremina filtrata u čaši zapremine 800 ml podesi se na približno 600 ml.
Rastvor treba da bude bezbojan i bistar.
Nitrati koji smetaju titraciji razaraju se sa 10 ml 20 %-nog rastvora NaHSO3. Rastvor se prokuva, doda mu se iz birete oko 30 ml koncentrovanog rastvora H3PO4, takođe se doda nekoliko kapi metil-oranža, nastavi se dodavanje H3PO4 do neutralizacije i, najzad, doda 1,5 ml H3PO4 u višku. Ukupna potrebna količina H3PO4 malo je manja od 35 ml, izuzev kad se proces vodi tako da je KNO3 bio redukovan većim količinama organske supstancije u K2CO3. Višak kiseline treba izbegavati jer daje niske rezultate. Kiselinu treba dodavati relativno brzo, jer se boja metiloranža vremenom gubi s obzirom na nepotpunu razgradnju nitrata.
Posle neutralisanja zapremina se upari kuvanjem, koje traje 20 min. Zatim se doda malo bromne vode i kuva dok rastvor ne postane trajno žut, kuvanje se nastavi do bezbojnosti, a zatim se kuva još 5 min. U rastvor se zatim doda nekoliko kapi salicilne kiseline da bi se odstranili poslednji tragovi broma, rastvor se ohladi, doda se 5 ml 20%-nog redukovanog rastvora H3PO4 i 0,5 do 1 g KJ. Rastvor se titrira 0,005 mol/l rastvorom Na2S2O3, uz dodavanje rastvora skroba. Zapremina rastvora po završetku titriranja treba da iznosi 400 do 500 ml.
Izračunavanje
Količina joda izražava se u miligramima, a izračunava se po sledećoj formuli:
f · a
gde je:
f - faktor za preračunavanje 0,005 mol/l rastvora Na2S2O3 (ml) u jodu (mg);
a - broj utrošenih ml 0,005 mol/l rastvora Na2S2O3.
33. Određivanje mikroelemenata - spektrofotometrijska metoda
Princip i primena
Za određivanje mikroelemenata: gvožđa, mangana, kobalta, cinka i bakra primenjuje se spektrofotometrijska metoda. To određivanje obavlja se za sve vrste stočne hrane.
Pripremanje uzorka
Pri uzimanju i sitnjenju uzorka za ispitivanje ne sme se koristiti oprema koja je po površini korozirala ili se naziru tragovi oštećenja.
Spaljivanje
Za određivanje mikroelemenata, uzorak za ispitivanje spaljuje se na temperaturi od 450 do 500°C, kako ne bi došlo do gubitaka ili do stvaranja nerastvorljivih jedinjenja sa kiseonikom iz vazduha ili sa zidovima posude u kojoj se vrši spaljivanje. Za spaljivanje se koriste platinske, kvarcne ili porculanske posude čija površina treba da bude glatka i glazirana.
Pripremanje rastvora za analizu
Odmeri se 10 g uzorka za analizu i spaljuje na temperaturi 450 do 500°C dok se ne dobije ostatak bez ugljenika (3 do 6 h).
Završetak spaljivanja određuje se prema boji pepela. Ostatak se kvantitativno prenese u čašu zapremine 600 ml. Uzorci koji nisu organskog porekla ne spaljuju se. Čaša se poklopi sahatnim staklom i pažljivo se doda koncentrovana hlorovodonična kiselina. Pomoću staklenog štapića napravi se kaša i drži na vodenom kupatilu dok sva kiselina ne ispari. Eventualni ostatak kiseline ukloni se na peščanom kupatilu.
Dobijenom ostatku u čaši doda se 60 ml koncentrovane hlorovodonične kiseline i 300 ml vode, pa se zatim namanje 30 min drži na ključalom vodenom kupatilu. Posle toga filtrira se preko gustog filtrir-papira u odmerni sud zapremine 500 ml. Čaša i filtrir-papir dobro se isperu vodom, a odmerni sud se do oznake dopuni vodom. Tako pripremljen rastvor koristi se za određivanje mikroelemenata.
34. Određivanje mangana - spektrofotometrijska metoda
Princip i primena
Metoda se zasniva na oksidaciji bezbojnog mangana Mg (II) u crvenoljubičasti obojeni mangan. Kao oksidaciono sredstvo koristi se perjodat ili persulfat, uz srebro-nitrat kao katalizator. Sumpornom i azotnom kiselinom odstranjuju se hloridi, redukujuće supstancije i male količine organske materije, a uticaj trovalentnog gvožđa odstranjuje se dodavanjem ortofosforne kiseline.
U mineralnim smešama u kojima su zastupljene velike količine hlorida koristi se perjodat kao oksidaciono sredstvo.
Reagensi
Za određivnje sadržaja mangana potrebni su sledeći reagensi:
1) standardni rastvor MnSO4H2O: rastvori se 0,3077 g MnSO4H2O u 0,1 mol (1/2 H2SO4) /l i do 1000 ml dopuni sumpornom kiselinom iste koncentracije:
1 ml = 0,1 mg Mn;
2) rastvor sumporne kiseline 5 : 100 (V/V).
U vrlo malim količinama mangana koncentracija sumporne kiseline treba da iznosi c(1/2H2SO4) = 1 mol/l, a za veći sadržaj mangana - koncentracija sumporne kiseline c(1/2H2SO4) = 1,75 mol/l;
3) koncentrovana sumporna kiselina (r = 1,84 g/ml);
4) koncentrovana azotna kiselina (r = 1,40 g/ml);
5) rastvor ortofosforne kiseline, 85%-ni;
6) kalijum-perjodat.
Napomena. |
Perjodat je pogodniji za određivanje mangana u mineralnim smešama. Nedostatak persulfata je u tome što stvara talog i kalcijum-sulfat, a može eventualno stvoriti koloidni srebro-hlorid. Crvena boja nastala oksidacijom persulfata gubi se posle 24 sata, a boja nastala oksidacijom perjodata postojana je mesec dana. |
Određivanje
U porculansku šolju odmeri se 25 do 100 ml rastvora pripremljenog za analizu (metoda 33 ovog pravilnika) - (sa sadržajem mangana 0,05 do 0,5 mg), doda se 2 ml koncentrovane sumporne kiseline i 1 ml koncentrovane azotne kiseline. Šolja se stavi na ključalo kupatilo da tečnost ispari i posle toga drži u sušnici na 140°C u toku jednog časa, dok se ne odstrani miris na kiseline.
Ostatku u šolji doda se 25 ml vode, zagreje se i filtrira kroz filtrir-papir u koničnu tikvicu zapremine 100 ml, a šolja i ostatak na filtrir-papiru tretiraju se sa 15 ml razblažene sumporne kiseline (5 : 100 V/V). Zatim se u koničnu tikvicu doda 2 ml 85 %-ne ortofosforne kiseline i oko 0,5 g kalijum-perjodata, pa se sadržaj zagreva do ključanja. Konična tikvica, poklopljena sahatnim staklom, ostavi se 1 h na vodenom kupatilu koje intenzivno ključa. Posle hlađenja, rastvor se prenese u odmernu tikvicu zapremine 50 ml i vodom dopuni do marke. Dobro se promeša i boja meri na 525 nm u odnosu na slepu probu.
Standardna kriva priprema se od standardnog rastvora MnSO4H2O, čiji 1 ml sadrži 0,1 mg Mn. Uzima se 0,5; 1,0; 2,0; 3,0; 4,0 i 5,0 ml, prenese u koničnu tikvicu, doda 2 ml sumporne kiseline, oko 2 ml vode, 2 ml ortofosforne kiseline i dalje postupa kao što se postupa sa rastvorom kome se meri boja na 525 nm.
35. Određivanje kobalta - spektrofotometrijska metoda
Princip i primena
Kobalt gradi sa 2-nitrozo-1-naftolom crveno obojeni kompleks koji se rastvara u organskim rastvaračima. Višak reagensa se odstranjuje mućkanjem ekstrakta toluola razblaženim natrijum-hidroksidom koji ne utiče na kobalt-nitrozo-komplekse. Trovalentno gvožđe, koje sa 2-nitrozonaftolom gradi mrk talog, maskira se citratom. Mineralne smeše koje sadrže bakar, koji takođe daje reakciju s nitrozonaftolom, mogu se analizirati do odnosa kobalt - bakar 1:150.
Reagensi
Za određivanje sadržaja kobalta potrebni su sledeći reagensi:
1) standardni rastvor kobalta: rastvori se 0,4769 g CoSO4 · 7H2O u 0,1 mol (HCl)/l i dopuni kiselinom do 1000 ml (1 ml = 0,1 mg Co). Od tog rastvora otpipetira se 10 ml u odmerni sud zapremine 500 ml i dopuni rastvorom 0,1 mol (HCl)/l do oznake (1 ml = 2 µg Co);
2) 1 %-ni rastvor 2-nitrozo-1-naftola u metanolu (m/V).
Usled stajanja rastvora, deo nitrozojedinjenja u prisustvu kiseonika iz vazduha prelazi u nitrojedinjenje i slepa proba daje visoke vrednosti, tako da pripremljeni rastvor uvek mora biti svež.
Osim toga, trgovački preparat nije dovoljno čist i treba ga prečistiti na taj način što se 10 g preparata rastvori u 400 ml ključale vode, ostavi da se ohladi i istaloženi kristali suše u vakuumu na temperaturi 70°C;
3) 50%-ni diamonijum-hidrogencitrat (m/V);
4) rastvor natrijum-hidroksida c(NaOH) = 2 mol/l;
5) toluol.
Određivanje
U tikvicu za jodni broj odmeri se 10 do 50 ml rastvora za analizu (metoda 33 ovog pravilnika), sa sadržajem CO do 50 µg, i doda vode do 50 ml. Rastvor se tretira sa 10 do 20 ml 50%-nog amonijumcitratnog rastvora, a pomoću rastvora 2 mol (NaOH)/l pH vrednost podesi na 6,7 do 6,8. Zatim se doda 1 ml 1 %-nog nitrozonaftola i rastvor ostavi 90 min na tamnom mestu. Doda se 20 ml toluola i promućka, a supspenzija odmah prenese u levak za odvajanje i snažno mućka 5 min. Posle odvajanja faza, ispusti se vodeni sloj, čep i zidovi operu vodom, ispusti se voda, a zatim doda 25 ml rastvora 2 mol (NaOH)/l i promućka. Posle toga se ispusti natrijum-hidroksid, čep i zidovi ponovo operu vodom, voda se ispusti i ekstrakcija hidroksidom ponovi snažnim mućkanjem 5 min. Posle ispuštanja vodenog sloja uzima se ekstrakt toluola u kivetu dužine 2 cm i spektrofotometriše u odnosu na slepu probu na 530 nm.
Za pripremanje standardne krive uzima se 2,5; 5,0; 7,5; 10,0; 15,0 i 20,0 ml standardnog rastvora, koji u mililitru sadrži 2 µg Co u tikvici za jodni broj, dopuni vodu do 50 ml i dalje postupa kao što je navedeno u postupku određivanja.
36. Određivanje gvožđa - spektrofotometrijska metoda
Princip
U alikvotnom delu rastvora pripremljenog za analizu (metoda 33 ovog pravilnika) trovalentno gvožđe se prevede u dvovalentno pomoću hidroksilamin-hlorhidrata, rastvoru se, pomoću natrijuma-acetata, pH vrednost podesi na 3,5 i sa 1,10 - fenantrolinom nagradi kompleks crvene boje, koji se fotometriše.
Reagensi
Za određivanje gvožđa koriste se sledeći reagensi:
1) 10 %-ni rastvor hidroksilamin-hlorhidrat: odmeri se 10 g NH2OH HCl, rastvori se u vodi i dopuni vodom do 100 ml. Čuva se u frižideru;
2) rastvor natrijum-acetata c(CH3COONa · 3H2O) = 4 mol/l : 544 g CH3COONa · 3H2O rastvori se u vodi i dopuni vodom do 1000 ml;
3) puferni rastvor: 3,5 ml 4 mol (CH3COONa · 3H2O)/l dopuni se razblaženom sirćetnom kiselinom do 100 ml (115 ml glacijalne sirćetne kiseline razblaži se vodom do 1 litra), a zatim razblaži vodom do 1000 ml;
4) 1,10 - fenantrolin: odmeri se 0,2 g 1,1 - fenantrolin-monohidrata i rastvori u 100 ml hladne vode. Rastvor treba da je bezbojan i da se čuva u frižideru;
5) rastvor hlorovodonične kiseline c(HCl) = 2 mol/l;
6) koncentrovana hlorovodonična kiselina (z = 1,19 g/ml);
7) standardni rastvor gvožđa rastvori se 0,8635 g NH4Fe (SO4)2 12 H2O u vodi, doda 100 ml koncentrovane hlorovodonične kiseline (r = 1,19 g/ml) i dopuni vodom do 1000 ml (1 ml odgovara 100 µg gvožđa). Od tog rastvora otpipetira se 50 ml u odmerni sud zapremine 250 ml i dopuni vodom do oznake (1 ml tako razblaženog rastvora odgovara 20 µg gvožđa i uvek se priprema neposredno pred upotrebu.
Određivanje
U odmerni sud zapremine 100 ml odmeri se alikvotni deo rastvora pripremljenog za analizu (metoda 33 ovog pravilnika), sa sadržajem gvožđa do 0,4 mg, doda rastvora 2 mol (HCl)/l tako da ukupna zapremina bude oko 30 ml, zatim se 1 ml hidroksilamin-hlorhidrata promućka i ostavi najmanje 2 min. Posle toga se pomoću 4 mol (CH3COONa · 3H2O)l, pH vrednost podesi na približno 3,5. Ako se rastvor zamuti zbog velike zastupljenosti kalcijuma u fosforu, doda se još nekoliko mililitara 2 mol (HCl)/l i pH vrednost ponovo podesi na 3,5. Zatim se dola 10 ml 1,10-fenantralina i puferom dopuni do oznake, promućka i ostavi 2 h.
Meri se ekstinkcija na 510 nm u kiveti dužine 1 cm u odnosu na slepu probu, koja sadrži samo reagens.
Za pripremanje standardne krive uzima se 0; 2,0; 5,0; 10,0; 15,0 i 20 ml razblaženog standardnog rastvora u odmerni sud zapremine 100 ml do 30 ml dopuni 2 mol (HCl)/l i postupa kao što je utvrđeno u postupku određivanja.
37. Određivanje cinka - spektrofotometrijska metoda
Princip
U neutralnom ili slabo alkalnom rastvoru cink sa ditizonom (difenil-tiokarbazon) gradi crveno obojeni kompleks koji se ekstrahuje hloroformom ili ugljen-tetrahloridom. Ekstrakcija ugljen-tetrahloridom brža je nego ekstrakcija hloroformom. Metoda određivanja preko mešane boje u odnosu na jednu boju jednostavnija je i tačnija, pa se cink najpre izoluje, što se postiže razlaganjem cink-ditizonata u razblaženoj hlorovodoničnoj kiselini, uz dodavanje natrijum-dietilditiokarbamata.
Reagensi
Za pripremanje rastvora moraju se koristiti bidestilovana voda i bidestilovani amonijak.
Osim toga koriste se i sledeći reagensi:
1) standardni rastvor ZnSO4 · 7H2O: odmeri se 0,4398 g ZnSO4 · 7H2O rastvori u 0,1 mol (HCl)/l i dopuni hlorovodoničnom kiselinom do 1000 ml (1 ml = 0,1 mg Zn). Od tog rastvora pipetom se odmeri 50 ml u odmernu tikvicu zapremine 500 ml i do oznake dopuni 0,1 mol (HCl)/l (1 ml = 0,01 mg Zn);
2) 0,01%-ni rastvor difenil - ditiokarbazona u ugljen tetrahloridu (m/V);
3) 10%-ni (m/V) ) rastvor diamonijum-hidrokarbazona, toliko dugo tretiran rastvorom 0,01 % (m/V) ditizona dok ugljen-tetrahloridni sloj ne postane zelen;
4) rastvor monohidrata limunske kiseline: rastvori se 210 g monohidrata limunske kiseline u približno 600 ml vode, doda se 350 ml 10 mol (NH3)/l i dopuni vodom do oznake (pH vrednost 9,4). Rastvor se čuva u polietilenskoj boci i postojan je sedam dana;
5) 0,25 %-ni rastvor natrijum-dietil-ditiokarbamata, koji se priprema neposredno pred upotrebu (m/V);
6) mešani rastvor: u 100 ml pripremljenog rastvora monohidrata limunske kiseline doda se oko 300 ml vode, 50 ml pripremljenog rastvora 0,25 % (m/V) natrijum-dietil-ditiokarbamata i dopuni vodom do 500 ml;
7) bidestilovan rastvor hlorovodonične kiseline, c(HCl) = 0,1 mol/l;
8) bidestilovan rastvor hlorovodonične kiseline, c(HCl) = 0,02 mol/l;
9) 0,02 %-ni rastvor fenolftaleina u etalonu;
10) ugljen-tetrahlorid;
11) 0,01 %-ni rastvor ditizona.
Određivanje
Alikvotni deo pripremljenog rastvora za analizu (metoda 33 ovog pravilnika) koji sadrži do 80 µg cinka uzima se u levak za odvajanje i dopuni vodom do 20 ml, zatim se doda 5 ml 10 %-nog amonijum-citrata (ako se ras-tvor zamuti doda se još amonijum-citrata dok se zamućenje ne izgubi) i tri kapi fenolftaleina i koncentrovanog amonijaka sve dok boja ne počne da se menja. Posle dodavanja dve kapi amonijaka u višku i oko 5 ml rastvora 0,01%-nog ditizona, rastvor se dva minuta snažno mućka (najbolje na mućkalici) i ekstrakt ditizona prenese u drugi levak za odvajanje. Mućkanje ditizonom ponovi se na isti način toliko puta dok boja ne postane zelena.
Preporučuje se da se posle svakog izdvajanja ekstrakta ditizona zidovi levka za odvajanje isperu sa 2 ml čistog ugljen-tetrahlorida kako bi se pokupile kapi koje se nalaze na površini (ne treba mućkati).
Sjedinjeni ekstrakti ditizona snažno se mućkaju 2 min sa 50 ml 0,02 ml (HCl)/l, pri čemu cink prelazi u vodenu fazu. Rastvor ditizona se baci, a rastvor hlorovodonične kiseline se još jedanput kratko izmućka sa 5 do 10 ml čistog ugljen-tetrahlorida. U kiseli rastvor hlorovodonične kiseline doda se 50 ml mešanog rastvora i 10 ml 0,01% rastvora ditizona. Smeša se, 1 min snažno mućka i ekstrakt ditizona se filtrira kroz filtrir-papir (bela traka 7 cm) u odmerni sud zapremine 50 ml. Tada se gornja površina vodenog rastvora ispere sa 2 ml čistog ugljen-tetrahlorida i bez mućkanja ispusti u odmerni sud. Posle ispiranja filtrir-papira doda se ugljen-tetrahlorid do oznake. Rastvor se promućka i boja meri na 535 nm u odnosu na slepu probu, u kiveti dužnie 0,5 cm.
Za pripremanje standardne krive uzima se 1 ml standardnog rastvora koji odgovara 0,01 mg Zn. Uzima se 0; 1,0; 2,0; 4,0; 6,0 i 8,0 ml, dopuni se vodom do 20 ml i dalje postupa kao što je u utvrđeno u postupku određivanja.
38. Određivanje bakra - spektrofotometrijska metoda
Princip
Kao reagens za određivanje koristi se olovna so dietil-ditiokarbamat, na koju nemaju uticaja joni Fe (III), Mn(II), Ni(II) i Co(II).
Smetnje pričinjavaju srebro, živa i talijum, ali kad su zastupljeni dvadeset puta više od bakra. Već 3 µg bizmuta smetaju određivanju 10 µg bakra, pa se prisutan bizmut tada odstranjuje mućkanjem organskog ekstrakta sa rastvorom 5 mol (HCl)/l ili rastvorom 2 mol (NaOH)/l. Bakar-dietil-ditiokarbamat kompleks može se ekstrahovati toluolom, hloroformom ili ugljen-tetrahloridom.
Reagensi
Za određivanje bakra koriste se sledeći reagensi:
1) standardni rastvor bakra: odmeri se 0,3928 g CuSO4 · 5H2O rastvori u 1 mol (HCl)/l i dopuni hlorovodoničnom kiselinom do 1000 ml (1 mol = 0,1 mg Cu). Od tog rastvora uzima se 20 ml i vodom razblaži do 1 litra (1 ml = 2 ng Cu);
2) 0,01%-ni rastvor olovo-dietilditiokarbamata u toluolu. Rastvor se čuva u mraku ili u tamnoj boci;
3) 50%-ni rastvor diamonijum-hidrogencitrata (m/V);
4) rastvor natrijum-hidroksida c(NaOH) = mol/l;
5) bidestilisan amonijak;
6) toluol;
7) 1%-ni rastvor fenolftaleina u etanolu (m/V).
Određivanje
Odmeri se 1 do 10 ml rastvora pripremljenog za analizu (metoda 33 ovog pravilnika) koji sadrži do 40 µg bakra i prenese u levak za odvajanje, zapremine 100 ml, dopuni sa 20 ml vode, doda 5 ml 50%-nog diamonijum-citrata i 1 do 2 kapi fenolftaleina i amonijaka, sve dok boja ne počne da se menja. Zatim se doda 20 ml olovo-dietil-ditiokarbamata, mućka 5 min, ostavi se da se faze razdvoje i ispusti se vodeni sloj. Čep i zidovi levka operu se vodom. Organska faza se tretira sa 25 ml rastvora 2 mol (NaOH)/l i mućkanje ponovi 5 min. Posle odvajanja faza ispusti se vodeni sloj, a ekstrakt toluola se preko filtrir-papira (bela traka) ispusti u odmerni sud zapremine 25 ml i toluolom dopuni do oznake. Zatim se u kiveti dužine 2 cm meri apsorbancija žuto obojenog kompleksa na 435 nm u odnosu na slepu probu.
Standardna kriva priprema se od razblaženog standardnog rastvora koji u 1 ml sadrži 2 µg bakra. U levak za odvajanje uzima se 2,5; 5,0; 10,0; 15,0 i 20 ml razblaženog standardnog rastvora i dalje postupa kao što je utvrđeno u postupku određivanja.
39. Određivanje karotina (brzi postupak)
Princip i primena
Princip se zasniva na ekstrakciji pigmenta u petroletru i odvajnju pigmenata primenom kolonske hromatografije i određivanju intenziteta boje spektrofotometrijskim merenjem.
Metoda se primenjuje za brzo određivanje karotina u svežim i suvim uzorcima stočne hrane.
Reagensi
Potrebni su sledeći reagensi, čistoće pro analysi, a voda mora biti destilovana:
1) petroletar, tačke ključanja između 30° i 70°C;
2) petroletar, tačke ključanja između 80° i 120°C;
3) ekstrakciona smesa, koja se sastoji iz jedne zapremine petroletra, tačke ključanja između 40° i 70°C i dve zapremine petroletra, tačke ključanja između 80° i 120°C;
4) aceton;
5) natrujum-sulfat, bezvodni;
6) apsorbens: komercijalno koštano brašno ekstrahovano 8 h acetonom i 8 h petroletrom, zatim sušeno i prosejano kroz sito sa otvorima čiji je prečnik 0,36 mm.
Aparatura i pribor
Osim uobičajene laboratorijske opreme, potreban je sledeći pribor:
1) vodeno kupatilo, sa regulacijom temperature na 90°C;
2) hromatografska cev, unutrašnjeg prečnika 12,5 mm i dužine 30 cm, sa kapilarnom cevi čiji je donji deo zaronjen u grlo odmerne tikvice zapremine 100 ml;
3) vakuumska naprava za filtraciju za hvatanje eluata u odmernu tikvicu (videti sl. 4 u prilogu);
4) analitička vaga.
Pripremanje svežeg biljnog uzorka iz silaže
Odmeri se 100 g dobro promešanog uzorka i isecka nožem ili odgovarajućom sečkom.
Pripremanje uzorka iz suve biljne mase
Samelje se 20 g suve biljne mase, tako da usitnjene čestice mogu proći kroz sito veličine otvora 1 mm.
Određivanje
Ekstrakcija pigmenata
Ekstrakcija pigmenata iz sveže biljne mase i silaže
Odmeri se 2 g pripremljenog uzorka sa tačnošću 0,001 g, stavi se u tarionik, doda se 5 g peska i 5 g bezvodnog natrijum-sulfata i meša dok se ne dobije suv homogeni prašak.
Samlevena masa se kroz cev ubaci u mernu posudu zapremine 100 ml. Tarionik se ispere postepenim dolivanjem u bocu 20 ml ekstrakcione smeše. Povratni kondenzator se namesti na bocu, koja se uroni u vodeno kupatilo zagrejano na 90°C i ostavi 1 h.
Ekstrakcija pigmenata iz suve biljne mase
U mernu bocu zapremine 100 ml odmeri se 1 g uzorka, sa tačnošću 0,01 g, i doda se 40 ml ekstrakcione smeše. Povratni kondenzator se namesti na bocu, koja se uroni u vodeno kupatilo zagrejano na 90°C i ostavi 1 h.
Odvajanje pigmenata
Hromatografska kolona u hromatografskoj cevi napuni se apsorbensom. Pre punjenja kolone, na donji deo cevi stavi se malo staklene vune ili pamuka. Visina sloja apsorbensa mora iznositi oko 10 cm. Slaganje apsorbansa reguliše se pomoću vakuumske sisaljke, gornja površina apsorbensa se poravna tupim štapićem i na nju stavi sloj bezvodnog natrijum-sulfata visine 1 cm i čvrsto pritisne. Hromatografska kolona se navlaži petroletrom, čija je tačka ključanja između 40° i 70°C. Za regulisanje protoka ekstrakta u koloni koristi se vakuum i eluat hvata u odmernu tikvicu zapremine 100 ml. Za eluiranje karotina upotrebljava se 50 ml petroletra, tačke ključanja između 40° i 70°C, u nekoliko navrata, tako da površina kolone nikad ne bude bez tečnosti. Dovoljno je 50 ml da karotinski obruč kvantitativno pređe u eluat. Ta hromatografska kolona se upotrebljava sve dok se ne dobije zona ostalih pigmenata do 2 cm iznad donjeg dela cevi. Eluat se do oznake dopuni petroletrom, tačke ključanja između 40° i 70°C i spektrofotometrom se odredi sadržaj karotina.
Određivanje apsorbancije
Apsorbancija rastvora određuje se pomoću spektrofotometra, na talasnoj dužini od 450 nm i pripremi se standardna prava iz standardnih rastvora karotina visoke čistoće. Za veću tačnost treba napraviti takve koncentracije rastvora da njihova apsorbancija bude između 0,25 i 0,75.
Izračunavanje
Sadržaj karotina izražava se u mg/kg i izračunava se po sledećoj formuli:
m · V |
||
m1 · l |
gde je:
m - karotin očitan sa krive, u µg;
V - zapremina eluata, u mililitrima;
m1 - odvaga uzorka u eluatu, u µg;
l - debljina kivete, u centimetrima.
Ako ne postoji standardna kriva, karotin se izračunava po sledećoj formuli:
E · V · 100 |
||
260 · l · m1 |
gde je:
E - apsorbancija eluata;
V - zapremina eluata, u mililitrima;
m1 - masa uzorka u eluatu, u µg;
l - debljina kivete, u centimetrima.
Ponovljivost
Razlika između rezultata dva određivanja na istom uzorku koja jedno za drugim izvodi isti analitičar ne sme biti veća od 8% srednje vrednosti dobijenog rezutata.
40. Određivanje karotina i ksantofila u svežim i suvim uzorcima biljnog porekla
Princip i primena
Metoda se primenjuje za određivanje karotina i ksantofila u uzorcima svežeg i suvog biljnog materijala, koji sadrži više od 30 mg karotina po 1 kg stočne hrane.
Princip se zasniva na ekstrakciji suve biljne mase pomoću heksana i acetona, a sveže hrane - na ekstrakciji acetonom. Posle ekstrakcije vrši se osapunjenje pomoću alkoholnog rastvora kalijum-hidroksida, ekstrakt pigmenta se uparava, ostatak se rastvara u petroletru i karotin hromatografski odvoji eluiranjem petroletrom, a ksantofil - eluiranjem etanolom. Količina izdvojenih pigmenata određuje se spektrofotometrijskim merenjem.
Reagensi
Koriste se sledeći reagensi, čistoće pro analysi i destilovana voda:
1) n-heksan, čist;
2) aceton, čist;
3) n-heksan-acetonska smeša (7:3),70 delova čistog n-heksana i 30 delova čistog acetona;
4) petroletar (tačka ključenja 30 do 50°C);
5) 40 %-ni etanolni rastvor KOH; pre upotrebe rastvori se 4 g kalijum-hidroksida u nekoliko kapi vode i do 10 ml dopuni etanolom;
6) natrijum-sulfat, bezvodni;
7) natrijum-sulfat, zasićen vodeni rastvor;
8) aluminijum-oksid za hromatografiju, standardizovan po Brokmanu (Brockman), žari se 8 h na temperaturi 750°C, ohladi u eksikatoru i čuva u boci od tamnog stakla sa brušenim zatvaračem.
Pre upotrebe za hromatografiju doda se 91 g aluminijum-oksida i 9 ml destilovane vode, snažno promućka i 9 h čuva u tamnoj boci sa brušenim zatvaračem;
9) etanol, 96 %-ni;
10) azot, gas.
Aparatura i pribor
Osim uobičajene laboratorijske opreme, koristi se i sledeći pribor:
1) kolone za hromatografiju, dužine od 15 do 30 cm, prečnika od 10 do 15 mm;
2) vakuum-uparivač;
3) spektrofotometar.
Pripremanje uzorka iz svežeg biljnog materijala
Odmeri se 10 g iseckanog biljnog materijala, sa tačnošću od 0,001 g, stavi se u homogenizator, doda 50 ml acetona i meša tako da se pigmenti ekstrahuju. To se ponovi najmanje tri puta. Sjedinjeni ekstrakti se filtriraju preko staklenog filtrir-lončića u mernu tikvicu zapremine 200 ml i tikvica se dopuni acetonom do oznake.
Pripremanje uzorka iz suvog biljnog materijala
U odmerni sud odgovarajuće zapremine ili u odgovarajuću bocu sa brušenim zatvaračem odmeri se od 2 do 10 g, sa tačnošću od 0,001 g, fino usitnjenog uzorka prosejanog kroz sito veličine otvora 0,5 mm, doda od 30 do 50 ml smeše heksana i acetona, uvede azot ili drugi inertni gas, promućka i ostavi na tamnom mestu da prenoći.
Određivanje
Osapunjenje ekstrakta svežeg biljnog materijala
U levak za odvajanje otpipetira se od 20 do 50 ml ekstrakta, zavisno od očekivane količine pigmenta, doda se 0,5 ml 40 %-nog rastvora etanol-hidroksida i snažno promućka i ostavi da miruje pola sata. Zatim se doda od 30 do 50 ml petroletra i snažno promućka da bi pigmenti kvantitativno prešli u fazu petroletra. Donji sloj se ispusti, ostaci rastvora etanolnog kalijum-hidroksida i acetona se odstrane ispiranjem ekstraktom petroletra, dva puta sa po 100 ml vode. Na kraju se ostaci vode odstrane mućkanjem ekstrakta sa po 10 ml zasićenog vodenog rastvora natrijum-sulfata.
Prečišćeni ekstrakt petroletra blagim zagrevanjem u vakuumu uparava se do suvog ostatka i odmah rastvori sa 5 ml heksana.
Ako uzorak ne sadrži masti i hlorofil, saponifikacija se može izostaviti.
Osapunjenje ekstrakta suvog biljnog materijala
Jedan sat pre hromatografskog odvajanja pigmenata, u ekstrakt se doda 2 ml 40 %-nog etanolnog rastvora kalijum-hidroksida, snažno promućka i ostavi pola sata na tamnom mestu. Zatim se doda 2 ml destilovane vode, još jedanput promućka i ostavi da se slegnu čvrste čestice. Zatim se zapremina reguliše dodavanjem čistog n-heksana do oznake. Alikvotni deo, obično 50 ml, otpipetira se u bocu za vakuumsko uparavanje. Uparavanje se vrši do suva, blagim zagrevanjem do temperature 40°C i ostatak odmah rastvori sa 5 ml n-heksana.
Hromatografsko određivanje
Hromatografska cev se napuni aktiviranim aluminijum-oksidom. Pre punjenja kolone, na donji deo se stavi malo staklene vune ili pamuka. Visina sloja apsorbensa mora iznositi 10 cm. Na gornji sloj apsorbensa doda se sloj bezvodnog natrijum-sulfata, visine 2 cm, dobro ovlaži petroletrom i počinje hromotografija. Protok se reguliše vakuumom ili natpritiskom, tako da protok bude 1 do 2 kapi u sekundi. Karotin se eluira pomoću petroletra i eluat hvata u mernu bocu zapremine 50 ili 100 ml. Eluiranje je završeno kad se primeti da kapi više ne sadrže obojene materije. Eluat se u odmernoj boci dopuni petroletrom.
Ksantofil se eluira iz kolone pomoću 96 %-nog etanola, hvata se u mernu bocu zapremine 100 ili 200 ml, sve dok se ne dobiju bezbojne kapi. Zapremina se reguliše dodavanjem etanola do oznake.
Apsorbancija dobijenih eluata meri se na talasnoj dužini od 450 nm, uz obaveznu primenu standardne prave, i to: karotin prema petroletru, a ksantofil prema etanolu.
Izračunavanje
Količina karotina i ksantofila izražava se u mg/kg i izračunava se po sledećoj formuli:
očitavanje · B |
||
m · l |
gde je:
očitavanje - karotina, odnosno ksantofila sa standardne krive, u µg;
V - zapremina eluata, u mililitrima;
m - masa uzorka u eluatu (V), u mililitrima;
l - debljina optičkog sloja kivete, u centimetrima.
Ponovljivost
Razlika između rezultata dva uporedna određivanja koja je, istovremeno ili neposredno jedno za drugim, sa istim reagensima izveo isti analitičar, ne sme biti veća od 9 % od srednje vrednosti dobijenih rezultata.
Princip
Podesna količina uzorka osapunjuje se alkoholnim rastvorom KOH. Neosapunjivi deo, koji sadrži vitamin A, ekstrahuje se petroletrom, dobijeni ekstrakt čisti se preko kolone različitim apsorbensima, a u prečišćenom ekstraktu odredi se vitamin A Carr-Priceovim reagensom.
Reagensi
Za određivanje vitamina A koriste se sledeći reagensi:
1) etanol 99 %, p.a.;
2) kalijum-hidroksid, KOH, p.a.;
3) petroletar, tačke ključanja od 40 - 70°C, p.a.;
4) aceton, p.a.;
5) natrijum-sulfat, Na2S2O3, bezvodni, p.a.;
6) magnezijum-oksid, MgO, p.a.;
7) kiselgur G, za tankoslojnu hromatografiju;
8) aluminijum-oksid, Al2O3 - standard za hromatografsku adsorpcijsku analizu po Brokmanu, st. aktivnosti II-III;
9) hloroform, p.a.;
10) antimon-trihlorid, SvCl3 kristalni;
11) anhidrid sirćetne kiseline, p.a.;
12) vitamin A standard: kao standard se obično upotrebljava kristalizovan vitamin A acetat i vitamin A palmitat, rastvoren u ulju poznate jačine. Standard vitamin A palmitat rastvori se u prečišćenom hloroformu i razredi na koncentraciju od 30 I.J./ 1 ml - radni rastvor standarda.
Standard vitamin A acetat podvrgne se saponifikaciji i ekstrakciji (merenju), tako da koncentracija koja se meri u hloroformnom rastvoru bude 30 I.J./ 1 ml radnog rastvora standarda. Rastvor standarda priprema se neposredno pred upotrebu.
Aparatura i pribor
Za određivanje vitamina A koristi se sledeća aparatura i pribor:
1) aparatura za destilaciju u vakuumu NB 29, s tikvicama za otparavanje, zapremine 100, 250 i 500 ml;
2) tikvice za jodni broj, zapremime 100, 250 i 500 ml;
3) uređaj za hromatografiju: hromatografska kolona, dužine 20 cm, prečnika 2,50 cm (NB 14), tikvice sa okruglim dnom zapremine 100 ml NB 14, nastavak za hromatografiju na koloni NB 14;
4) vakuum-sisaljka;
5) kolorimetar ili spektrofotometar.
Pripremanje Carr-Priceovog reagensa
Masa od 120 g SbCl3, promeša se u tikvici za jodni broj sa približno 200 ml hloroforma, prethodno izmućkanog s vodom i predestilovanog uz korišćenje Vigreuhove kolone. Posle stajanja u tami, sadržina tikvice se zagreje do vrenja, dok se ne otopi sav SbCl3 i posle hlađenja prenese u suvu odmernu tikvicu zapremine 500 ml i dopuni do oznake. Reagens je upotrebljiv posle dva do tri dana.
Pripremanje kolone sa magnezijum-oksidom + kiselgurom
Magnezijum-oksid i kiselgur suše se 2 h pri temperaturi od 105°C, ohlade u eksikatoru i promešaju u odnosu 1 + 1 (m/m). Masa od 5 g te mešavine stavi se na kolonu, na čijem dnu se nalazi komadić vate. Kolona se puni pomoću vakuuma. Na tako pripremljenu kolonu uzorak se nanosi direktno, bez prethodnog ispiranja rastvaračem.
Pripremanje kolone sa aluminijum-trioksidom
Aluminijum-oksid se 3 h žari pri temperaturi 550°C i zatim upotrebljava kao adsorbens. Masa od 10 g tako pripremljenog Al2O3 promeša se sa 0,15 ml vode i stavlja na kolonu, na čijem se dnu nalazi komadić vate. Sloj bezvodnog natrijum-sulfata debljine 0,2 cm, stavi se na vrh kolone. Pre nanošenja uzorka, kolona se ispira petroletrom.
Određivanje
Homogenizovani uzorak u količini od 10 do 25 g, izmeren na analitičkoj vagi (količina zavisi od vrste stočne hrane, odnosno od koncentracije vitamina A u hrani), saponifikuje se uz dodavanje 150 ml etanola i 37,50 ml 50 %-nog rastvora KOH u etanolu. Saponifikacija se vrši uz povratno hlađenje 30 min. U levak za odeljivanje prebaci se 75 ml tekućeg sloja, doda se 60 ml vode i ekstrakcija vitamina A vrši sa 5 · 50 ml petroletra. Sakupljeni petroletarni ekstrakti peru se sa 3 · 50 ml vode i suše s natrijum-sulfatom, a zatim ispare do volumena od približno 20 ml. Ostatak se prebaci u suvu odmernu tikvicu zapremine 100 ml i sadržaj dopuni petroletrom do oznake. Količina od 50 ml petroletarskog ekstrakta, koja sadrži vitamin A i karotine, prebaci se na kolonu sa MgO + kiselgur, a eluiranje se vrši sa smesom rastvora acetona i petroletra u odnosu 2 + 98 (V/V)-(eluiranje karotina). Vitamin A se eluira sa smesom rastvora etanola i petroletra u odnosu 8 + 92 (V/V) dok sav vitamin ne bude eluiran. Sakupljeni eluati ispare do suvog ostatka u vakuumu na vodenom kupatilu pri temperaturi 60°C, a suvi ostatak se rastvori u 2 ml hloroforma.
Merenje
Koncentracija vitamina A meri se na kolorimetru, uz crveni filtar, ili na spektrofotometru, na talasnoj dužini 620 nm. Uporedo se meri apsorbancija uzorka i standardnog rastvora, uz slepu probu.
Slepa proba: 1 ml hloroforma, 6 kapi anhidrida sirćetne kiseline i 4 ml Carr-Priceovog reagensa.
Standard vitamina A : 0,20 ml hloroformnog rastvora standarda (6 I. J.) vitamina A, 0,80 ml hloroforma, 6 kapi anhidrida sirćetne kiseline i 4 ml Carr-Priceovog reagensa.
Uzorak: 0,2 ml hloroformnog rastvora uzorka, 0,80 ml hloroforma, 6 kapi anhidrida sirćetne kiseline i 4 ml Carr-Priceovog reagensa.
Zbog nestabilnosti plave boje, potrebno je odmah posle dodavanja Carr-Priceovog reagensa (najviše 10 sekundi) očitati apsorbanciju plave boje.
Izračunavanje
Koncentracija uzorka izražava se u mg/kg i izračunava po sledećoj formuli:
Ku = |
Eu · Ks · F · 1000 |
|
Es · m |
gde je:
Eu = |
ekstinkcija uzorka; |
Ks = |
koncentracija standarda (u I.J) |
Es = |
ekstinkcija standarda; |
F = |
faktor razređenja; |
m = |
odvaga uzorka, u gramima. |
A) Uzorak koji sadrži više od 50 mg/kg vitamina E
Princip
Uzorak koji se ispituje, saponifikuje se rastvorom KOH u etanolu, uz dodatak hidrokinona. Posle hlađenja reakcijske smese, ekstrahuje se alfa-tokoferol dietil-etrom, ekstrakt etra se ispira bazom, a zatim vodom i dopuni do određene zapremine. Alikvotni deo ekstrakta etra ispari u vakuumu do suvog ostatka, koji se otopi u benzenu i prebaci na prethodno pripremljenu kolonu hromatografa. Eluat ispari u vakuumu, a suvi ostatak se otopi u etanolu, čija je zapremina tačno određena. Alikvotni deo alkoholnog rastvora oksiduje se s rastvorom ferihlorida, doda se rastvor dipiridina i razvijena boja posle 2 min očita na fotometru na 520 mm, uz slepu probu. Na isti način i pod istim uslovima rada istovremeno se izmeri apsorbancija standarda poznate koncentracije. Iz podataka dobijenih merenjem i faktora razređenja izračuna se koncentracija vitamina E u uzorku.
Reagensi
Za određivanje vitamina E koriste se sledeći reagensi:
1) etanol 99 %-ni, p.a.;
2) hidrokinon, p.a.;
3) dietiletar, p.a.;
4) kalijum-hidroksid, KOH;
5) natrijum-sulfat, Na2S2O3, bezvodni, p.a.;
6) benzen, p.a.;
7) ferihlorid, FeCl3 : 50 mg FeCl3 · 6H2O rastvori se u 25 ml etanola (rastvor se priprema svež, svakodnevno);
8) aa´ - dipiridin: 50 mg otopi se u 10 ml etanola (rastvor se priprema svež, svakodnevno);
9) aktivna zemlja: 2 g Bleicherde prokuva se dva minuta s rastvorom, koji se priprema rastvaranjem 0,40 g SnCl2 · 2H2O i 10 ml koncentrovane HCl;
10) hlorovodonična kiselina, koncentrovana;
11) kieselgel G: 2 g kieselgela G, sušenog 30 min pri temperaturi 110°C i pomeša se s malo suvog benzena;
12) d,l-a - tokoferol standard: C29H50O2 čuva se na tamnom mestu, na sobnoj temperaturi, uz obavezno uvođenje inertnog gasa posle svake upotrebe.
Aparatura i pribor
Za određivanje vitamina E koristi se sledeća aparatura i pribor:
1) aparatura za destilaciju u vakuumu (NB), s tikvicama za isparavanje, zapremine 100, 250 i 500 ml;
2) tikvice za jodni broj, zapremine 300 ml;
3) levci za odeljivanje, zapremine 500 ml;
4) odmerne tikvice, čaše, obični levci;
5) uređaj za hromatografiju, hromatografska kolona dužine 20 cm, prečnika 2,50 cm (NB 14), tikvice okruglog dna, zapremine 100 ml (NB 14), nastavak za hromatografiju na koloni NB 14;
6) kolorimetar ili spektrofotometar;
7) vakuum-sisaljka.
Pripremanje kolone
Dva grama aktivne zemlje stavi se u hromatografsku kolonu, tečnost se ispusti, a adsorbens se tri puta ispere sa po 3 ml etanola. Ispiranje se nastavlja s benzenom (5 · 5 ml). Na kolonu se zatim stavi 2 g kiselgela, razmućenog u benzenu. Suvišak benzena ispusti se iz kolone, tako da nivo tečnosti bude 2 do 3 ml iznad granice slegnutog adsorbensa.
Pripremanje standarda vitamina E
U čistu i suvu odmernu tikvicu zapremine 25 ml odmeri se određena količina standarda vitamina E i, dodatnim razređivanjima etanolom, koncentrcija podesi na vrednost od približno 20 µg/2 ml. Standard vitamina E priprema se za svako određivanje vitamina E.
Postupak
Na analitičkoj vagi odmeri 1 g homogenizovanog uzorka u tikvicu za saponifikaciju zapremine 250 ml, doda se 30 ml etanola i 3 ml rastvora KOH 1 + 2 (V/V) i oko 50 mg hidrokinona. Saponifikacija se vrši na vodenom kupatilu, uz povratno hlađenje 30 min. Ovaj postupak, kao i svi kasniji postupci, izvodi se na mestu zaštićenom od svetlosti. Posle saponifikacije sadržaj tikvice se kvantitativno prebaci u levak za odvajanje, sa ukupno 50 ml destilovane vode. Tikvica se nekoliko puta ispere sa 50 ml etra, koji se takođe doda u levak za odeljivanje. Sadržaj u levku ekstrahuje se sa 5 · 50 ml etra. Sakupljeni ekstrakt etra ispira se vodom, a rastvor KOH u 3 %-nom etanolu i ponovo vodom, sve dok ekstrakt prestane da pokazuje baznu reakciju. Ostatak ekstrakta etra osuši se bezvodnim natrijum-sulfatom i prebaci u tikvicu za isparavanje uz dodavanje etra, kojim se ispira levak za odvajanje. Ekstrakt etra ispari u vakuumu do suvog ostatka na vodenom kupatilu pri temperaturi 60°C. U suvi ostatak doda se 5 ml benzena. Rastvor benzena prebaci se kvantitativno na kolonu i vitamin E eluira s ukupno 7 · 5 ml benzena. Sakupljeni eluati ispare u vakuumu do suvog ostatka pri temperaturi 60°C, a suvi ostatak se rastvori u 10 ml apsolutnog alkohola. Merenje se vrši posle 2 min, kako uzorka tako i standarda vitamina E, i to na fotoelektričnom kolorimetru uz zeleni filter, odnosno na spektrofotometru na 520 nm.
Merenje
Slepa proba: 3 ml alkohola + 1 ml ferihlorida + 1 ml aa´ - dipiridina
Standard vitamina E: 2 ml alkoholnog rastvora, koji sadrži oko 20 g vitamina E + 1 ml etanola + 1 ml ferihlorida + 1 ml aa´ - dipiridina
Uzorak: alikvotni deo od 2 ml alkoholnog rastvora vitamina E + 1 ml etanola + 1 ml aa´ - dipiridina + 1 ml ferihlorida.
Izračunavanje
Koncentracija uzorka izražava se u mg/kg vitamina E acetata i izračunava se po sledećoj formuli:
Ku = |
Eu · Ks · F · 1000 |
|
Es · m · 0,911 |
gde je:
Eu - apsorbancija uzorka;
Ks - koncentracija standarda, u miligramima;
Es - apsorbancija standarda;
F - faktor razređenja;
m - odmerni uzorak, u gramima;
0,911 - faktor za preračunavanje D, L alfa-tokoferola u vitamin E acetat.
B) Uzorak koji sadrži manje od 50 mg/kg vitamina E
Princip
Uzorak za ispitivanje saponifikuje se s rastvorom KOH u apsolutnom alkoholu (etanolu), uz dodavanje hidrokinona. Posle hlađenja reakcijske smese se ekstrahuju (alfa-tokoferol s dietil-etrom), ekstrakt etra ispira bazom, a zatim vodom i dopuni do određene zapremine. Alikvotni deo ekstrakta etra ispari u vakuumu do suvog ostatka koji se rastvori u apsolutnom alkoholu (etanolu). Alikvotni deo uzorka nanosi se na ploču za tankoslojnu hromatografiju (TLC) i ploča se stavi na razvijanje. Uz uzorak, na ploču se nanosi i standard tokoferola. Posle razvijanja, ploča se prska reagensom (ferihlorid + a - adipiridil). Mrlje rastvora uzorka i standarda moraju se međusobno podudarati po izgledu i po RF vrednostima.
Reagensi
Za određivanje vitamina E koriste se sledeći reagensi:
1) etanol 99 %, p.a;
2) dietil-etar p.a;
3) kalijum hidroksid, KOH;
4) natrijum sulfat, Na2S2O3, bezvodni;
5) hidrokinon, p.a;
6) benzen, p.a;
7) ferihlorid, FeCl3 : 50 FeCl3 x 6 H2O rastvori se u 25 ml etanola (rastvor se priprema neposredno pred upotrebu);
8) aa´ - dipiridin: 50 g otopi se u 10 ml etanola (rastvor se priprema neposredno pred upotrebu);
9) silikagel G za hromatografiju;
10) metanol, p.a.;
11) d.l - a - tokoferol standard C29H50O2 čuva se na sobnoj temperaturi, na tamnom mestu, uz obavezno uvođenje inertnog gasa posle svake upotrebe.
Aparatura i pribor
Za određivanje vitamina E koriste se sledeća aparatura i pribor:
1) aparatura za destilaciju u vakuumu (NB), s tikvicama za isparavanje, zapremine 250 ml;
2) tikvice za jodni broj;
3) levci za odeljivanje, zapremine 500 ml;
4) odmerne tikvice, čaše, obični levci;
5) uređaj za TLC;
6) staklene ploče, dimenzija 20 · 20 cm;
7) stakleni cilindri za razvijanje ploča za TLC, dimenzija 60 · 60 · 40 cm;
8) UV uređaj - fluorescentni ispitivač s kratkotalasnim i dugotalasnim svetlom (254 i 365 nm);
9) plastični most za podelu hromatografske podloge na polja i za nanošenje ekstrakta uzorka i rastvora standarda;
10) mikropipete, zapremine 10 µl.
Pripremanje ploča za TLC
Masa od 6 g silikagela G meri se u koničnoj tikvici sa staklenim čepom, zapremine 100 ml, i doda 20 ml mešavine rastvora 96 %-nog etanola i vode, u odnosu 135 + 15 (V/V). Ploča se očisti vatom natopljenom etanolom i sadržaj tikvice izlije se na tako očišćenu ploču. Tečan adsorbens iznivelira se tako da sloj bude jednoličan. Ploča se osuši na vazduhu, a zatim aktivira pri temperaturi 105°C u trajanju 1 do 2 h.
Pripremanje standarda vitamina E
U čistu i suvu odmernu tikvicu zapremine 25 ml odmeri se određena količina standarda vitamina E (alfa-tokoferol) i dodatnim razređenjima apsolutnim alkoholom koncentracija se podesi na vrednost 500 µg/l ml. Standard se priprema uz svako određivanje vitamina E.
Postupak
Na analitičkoj vagi odmeri se 10 g uzorka i prenese u tikvicu za saponifikaciju, zapremine 250 ml, doda 30 ml 99 %-nog etanola i oko 50 mg hidrokinona. Saponifikacija se vrši na vodenom kupatilu, uz povratno hlađenje 30 min. Ovaj postupak, kao i svi kasniji postupci, izvodi se na mestu zaštićenom od svetlosti. Posle saponifikacije, sadržaj tikvice se kvantitativno prebaci u levak za odeljivanje, sa ukupno 50 ml destilovane vode. Tikvica se nekoliko puta ispere sa ukupno 50 ml etra, koji se takođe doda u levak za odeljivanje. Sadržaj u levku se ekstrahuje sa 5 · 50 ml etra. Sakupljeni ekstrakti etra ispiraju se vodom, rastvorom KOH u 3 %-nom etanolu i ponovo vodom, dok ekstrakt prestane da pokazuje baznu reakciju. Ekstrakt etra osuši se bezvodnim natrijum-sulfatom i prebaci u tikvicu za isparavanje uz dodavanje etra, kojim se ispira levak za odeljivanje. Ekstrakt etra ispari u vakuumu do suvog ostatka na vodenom kupatilu, pri temperaturi 60°C. U suvi ostatak doda se toliko mililitara 99 %-nog etanola da u 30 ml bude 15 µg vitamina E (zavisno od količine vitamina E u uzorku). Na pripremljenu ploču za TLC nanosi se, u obliku crte (dužine 1,50 cm), 30 µl ekstrakta uzorka, 30 µl standarda i 30 µl S/2 (polovina koncentracije standarda, koja se dobija razređivanjem standarda s 99 %-nim etanolom, u odnosu 1 + 1). Tako pripremljena ploča stavlja se u cilindar za razvijanje, u kome se nalazi oko 200 ml razvijača (benzol i metanol u odnosu 1 : 99). Hromatogram se razvija dok front rastvarača dostigne visinu od približno 15 cm. Posle razvijanja, ploča se suši na vazduhu i prska reagensom: najpre rastvorom FeCl3, a zatim aa´ - dipiridilom. Mrlje rastvora uzorka i standarda moraju se međusobno podudarati po izgledu i po Rf-vrednostima. Koncentracija vitamina E očitava se upoređivanjem intenziteta obojenja ekstrakta uzorka s intenzitetom obojenja standardnog rastvora. Ako su intenziteti obojenja uzorka i standarda različiti, ponovi se TLC uz dodatna razređivanja ili standardnog rastvora ili rastvora uzorka. Kad intenzitet obojenja standardnog rastvora i uzorka postane identičan, izračuna se približna koncentracija uzorka, uz poznatu koncentraciju standarda i faktor razređenja.
Princip
Mikotoksini se određuju u tankoslojnom hromatografijom. Ohratoksin, kao relativno jaka kiselina, ekstrahuje se iz rastvora zasićenim natrijum-hidrokarbonatom; slabo kiseli zearalenon ekstrahuje se jakom bazom, tako da neutralni alfatoksin ostaje u rastvoru.
Reagensi
Za određivanje mikotoksina koriste se sledeći reagensi:
1) acetonitril - voda (90 + 10);
2) heksan;
3) hloroform, CHCl3;
4) zasićen rastvor NaHCO3 (pH = 7,8);
5) rastvor natrijum - hidroksida, c = 1 mol (NaOH)/l;
6) rastvor fosforne kiseline, c = 5 mol (1/3H3PO4)/l;
7) rastvor hlorovodonične kiseline, c = 1 mol (CHl)/l;
8) rastvor benzol-acetonitril (98 + 2);
9) bezvodni natrijum-sulfat, Na2SO4;
10) silikagel H, tip 60 za tankoslojnu hromatografiju;
11) rastvor toluola, etilacetata i mravlje kiseline (50 + 40 + 10);
12) rastvor sumporne kiseline i metanola (20 + 80);
13) standardni rastvori:
- alfatoksin B1 - 0,5 µg/ml;
- ohratoksin A - 5 µg/ml;
- zearalenon - 50 µg/ml;
- benzol-acetonitril (98 + 2).
Aparati
Koristi se aparatura za tankoslojnu hromatografiju.
Postupak
Ekstrakcija i separacija
U posudu zapremine 250 ml odmeri se 25 g fino samlevenog uzorka, doda 100 ml smeše acetonitrila i vode (90+ 10) i 10 min mućka u snažnoj mućkalici. Ekstrakt se filtrira preko filtrir-papira. Za dalju analizu koristi se 25 ml filtrata koji se obezmašćuje u levku za odvajanje, dva puta sa po 25 ml heksana. Ostavi se da se slojevi odvoje i heksanska faza ukloni, a acetonitrilska faza se ekstrahuje sa novih 25 ml heksana i ponovo se odstrani heksanska faza. Zatim se doda 25 ml vode i 8 ml zasićenog rastvora NaHCO3 i ekstrahuje sa 25 ml hloroforma. Donji sloj se ispusti u drugi levak za odvajanje i ponovo ekstrahuje sa 25 ml hloroforma. Celokupna hloroformska faza čini ekstrakt 1.
U levak za odvajanje, koji sadrži vodenu fazu, doda se 15 ml 1 mol (HCl)/l i ekstrahuje sa 20 ml hloroforma. Ekstrakcija kisele faze ponovi se sa još 20 ml hloroforma. Zatim se spojeni ekstrakt hloroforma filtrira kroz bezvodni Na2SO4 i koncentriše na 10 ml.
Koncentrisani ekstrakt prebaci se u malu staklenu posudu (zapremine oko 3 ml) uz hloroform i upari do suvog ostatka koji se otopi (ekstrakt 2) u 0,2 ml smeše benzol-acetonitrila (98 + 2) i ostavi za TLC.
U ekstrakt 1 doda se 10 ml 1 mol (NaOH)/L. Posle ekstrakcije ispusti se donji sloj u drugi levak za odvajanje i ekstrakcija ponovi sa istom količinom 1 mol (NaOH)/l. Pomešani ekstrakti natrijum-hidroksida su ekstrakti 1a.
Ekstrakt hloroforma ispere se sa 25 ml vode i donja faza se filtrira kroz bezvodni Na2SO4. Odvoji se vodena faza, hloroformski ekstrakt se prebaci u malu staklenu posudu i otopi u 0,2 ml rastvora benzol-acetonitrila (98 + 2) (ekstrakt 1b).
Ekstrakt 1a zakiseli se sa 8 ml 5 mol (1/3 H3PO4)/l i ekstrahuje dva puta sa 20ml hloroforma. Sakupljeni ekstrakti hloroforma filtriraju se kroz bezvodni Na2SO4, upare i otope u 0,2 ml smeše benzol-acetonitrila (98 + 2) i ostave za TLC.
Tankoslojna hromatografija
Ekstrakti 1a, 1b i 2, u količini od 5 do 10 ml, nanesu se na ploče dimenzije 20 · 20 cm prevučene slojem siliko-gela debljine 0,25 mm. Na iste ploče nanesu se i rastvori standarda mikotoksina, a zatim se ploče razvijaju u smeši rastvarača toluola, etilacetata i mravlje kiseline (50 + 40 + 10). Posle sušenja, mikotoksin se određuje upoređivanjem intenziteta fluorescencije mrlja uzorka i standarda.
U ekstraktu 1a nalazi se zearalenon, u ekstraktu 1b alfatoksin, a u ekstraktu 2 - ohratoksin.
Napomena. |
Po potrebi, mogu se sprovesti i testovi prskanjem 20 %-nom sumpornom kiselinom u metanolu i zagrevanjem 15 min na temperaturi 120°C ili dvodimenzionalnom hromatografijom. |
44. Određivanje aktivnosti antibiotika mikrobiološkim metodama na Petrijevim šoljama (metoda difuzije)
Princip
Aktivnosti antibiotika određuju se metodom difuzije na agarnoj podlozi, inokuliranoj odgovarajućim mikroorganizmom. Radi se na Petrijevim šoljama, s tim da se rastvori uzorka i standarda stavljaju u dodir s inokuliranom podlogom posredstvom metalnih cilindara. Referentna koncentracija standardnog niza upoređuje se s rastvorom uzorka, a taj rastvor mora sadržavati približno istu koncentraciju antibiotika kao i standard. Standardni niz sastoji se iz četiri koncentracije koje se pripremaju serijskim razređivanjima takozvanog radnog rastvora standarda, s tim da je jedna koncentracija u standardnom nizu referentna koncentracija.
Sva četiri rastvora standardnog niza stavljaju se u cilindre na svaku od ukupno 10 Petrijevih šolja. Rastvor uzorka stavlja se u cilindre na pet ploča, uz referentnu koncentraciju standarda, i to: u dva cilindra stavi se rastvor uzorka, a u druga dva - rastvor standarda. Posle korekcije srednjih vrednosti prečnika zona inhibicija dobijenih pojedinim rastvorima standardnog niza, nacrta se baždarni dijagram i očita korigovana srednja vrednost prečnika zona inhibicija za referentnu koncentraciju standardnog niza. Ta vrednost služi za korekciju srednje vrednosti prečnika zona inhibicija dobijenih rastvorom uzorka. Koncentracija antibiotika u uzorku očita se iz baždarnog dijagrama.
Sastav hranljivih podloga za određivanje antibiotika:
1) |
oksitetraciklin (CHTC) i hlortetraciklin (CHTC) pH vrednost posle sterilizacije = 5,6 - 5,7 |
|
|
pepton |
6,0 g |
|
ekstrakt kvasca |
3,0 g |
|
goveđi ekstrakt |
1,5 g |
|
agar |
15,0 g |
|
destilovana voda do |
1000 ml |
2) |
pepton |
6,0 g |
|
hidrolizovani kazein |
4,0 g |
|
ekstrakt kvasca |
3,0 g |
|
goveđi ekstrakt |
1,5 g |
|
dekstroza |
1,0 g |
|
agar |
15,0 g |
|
destilovana voda do |
1000 ml |
Posle sterilizacije, pH za oleandomicin, tilozin i neomicin iznosi 7,8 - 8,0, za benzatin-penicilin G 6,5 -6,6, a za virginiamicin 7,4. Za bacitracin iznosi takođe 6,5 - 6,6.
Sastav hranljivih podloga za uzgoj test-mikroorganizma (PODLOGA A):
pepton |
6,0 g |
hidrolizat kazeina |
4,0 g |
ekstrakt kvasca |
3,0 g |
goveđi ektrakt |
1,5 g |
dekstroza |
1,0 g |
agar |
15,0 g |
destilovana voda do 1000 ml, posle sterilizacije, pH vrednost iznosi 6,5 - 6,6. |
|
Puferi:
Koriste se sledeći puferi:
1) fosfatni pufer pH = 4,5: odmeri se 13,60 g KH2 PO4 u 1000 ml destilovane vode;
2) fosfatni pufer pH = 6: odmeri se 2,00 g K2 HPO4 i 8,00 g KH2 PO4 u 1000 ml destilovane vode;
3) fosfatni pufer pH = 8: odmeri se 16,73 g K2 HPO4 i 0,523 g KH2 PO4 u 1000 ml destilovane vode;
4) fosfatno-bikarbonatni pufer: pH = 8 odmeri se 16,73 g K2 HPO4, 0,523 g KH2 PO4 i 20,00 g NaHCO3 u 1000 ml destilovane vode.
Test-mikroorganizmi za određivanje antibiotika
1) BACILLUS CEREUS var. mycaides ATCC 9634 NCTC 10320
Bacillus sereus var. mycoides presađuje se jedanput mesečno na kosi agar (podloga A).
Pripremanje koncentrisanog inokuluma: 24 - časovna kultura gajena na kosom agaru (podloga A) pri temperaturi 37°C ispere se sa 5 ml sterilne destilovane vode, suspenzija prenese na Rouxevu bocu u kojoj se nalazi 300 ml podloge A i podjednako rasporedi po površini pomoću staklenih kuglica. Inkubacija traje sedam dana na temperaturi 37°C. Klice se suspenduju u 30 ml sterilne destilovane vode, suspenzija se prenese u sterilnu bocu, drži 30 min na temperaturi 65°C i centrifugira. Ispiranje spora vrši se tri puta sa po 10 ml sterilne destilovane vode i svaki put sterilno centrifugira. Spore se zatim resuspenduju u 30 ml sterilne destilovane vode, još jedanput greju 30 min na temperaturi 65°C, suspenzija se ohladi, sterilno homogenizuje i centrifugira 5 min na 1500 o/m. Istaloženi deo spora se baci, a ostatak suspenzije upotrebljava se kao inokulum (ako se čuva na temperaturi + 4°C, traje jednu godinu). U prethodnim probama se utvrdi zapremina nerazrađenog ili razrađenog inokuluma koji treba dodati određenoj količini hranljive podloge da se s najmanjom koncentracijom antibiotika u standardnom nizu dobiju dovoljno velike i oštre zone inhibicije.
2) SARCINA LUTEA ATCC 9341
Sarcina lutea ATCC 9341 presađuje se jedanput mesečno na kosi agar (podloga A): 24 - časovna kultura, gajena na kosom agaru, ispere se fiziološkim rastvorom (oko 3 ml). U prethodnim probama utvrdi se zapremina suspenzije inokuluma koji treba dodati određenoj količini hranljive podloge da se s najmanjom koncentracijom antibiotika u standardnom nizu dobiju dovoljno velike i oštre zone inhibicije. Sarcina lutea priprema se neposredno pred upotrebu. Taj mikroorganizam treba presaditi na kosi agar, isprati fiziološkim rastvorom, a zatim upotrebiti.
3) MICROCOCCUS PYOGENES VAR. AUREUS ATCC 6538 - P (Staphylococcus areus)
Micrococcus pyogenes var. aureus presađuje se dva puta mesečno na kosi agar (podloga A).
Pripremanje koncentrovanog inokuluma: 24-časovna kultura gajena na kosom agaru (podloga A) na temperaturi 32°C, ispere se sa 3 ml fiziološkog rastvora, suspenzija se prenese u Rouxovu bocu, koja sadrži 300 ml podloge A i 24 h inkubira na 32°C, a posle toga još 24 h na sobnoj temperaturi. Narasla kultura ispere se s 50 ml fiziološkog rastvora. Upotrebljiva je šest meseci na temperaturi + 4°C.
4) MICROCOCCUS FLAVUS ATCC 10240 - A NCTC 7743
Micrococcus flavus ATCC 10240 - A NCTC presađuje se dva puta mesečno na kosi agar (podloga A): 24 - časovna kultura, gajena na kosom agaru, ispere se fiziološkim rastvorom (oko 3 ml). U prethodnim probama se utvrdi zapremina suspenzije inokuluma koji treba dodati određenoj količini hranljive podloge da se s najmanjom koncentracijom antibiotika u standardnom nizu dobiju dovoljno velike i oštre zone inhibicije. Micrococcus flavus priprema se neposredno pred upotrebu, tj. taj mikroorganizam pre upotrebe treba presaditi na kosi agar, isprati fiziološkim rastvorom i zatim upotrebiti.
Aparatura i pribor
Osim uobičajene laboratorijske opreme, potrebne su i:
1) Petrijeve šolje (ploče), prečnika 100 mm (istih veličina i potpuno ravnog dna);
2) metalni cilindri izrađeni od nerđajućeg čelika, spoljnog prečnika 8 mm, unutrašnjeg prečnika 6 mm i visine 10 mm;
3) nivelisana ploča sa vijcima za regulaciju nagiba. Horizontalnost se proverava vodenom vagom;
4) graduisane pipete s ispusnim otvorom 3 do 4 mm za razlivanje podloge (podloga se mora otpipetirati u zdelice u roku od 2 do 3 sec);
5) šestar sa dva šiljka za merenje prečnika zona inhibicija. Merenje se vrši na milimetarskom papiru;
6) termostat;
7) mehanička električna mućkalica sa držačima za konične (erlenmajer) tikvice;
8) polulogaritamski papir za izradu baždarnog dijagrama.
Pripremanje Petrijevih šolja (ploča)
Petrijeve šolje sterilišu se na temperaturi 160°C. U prethodno zagrejane ploče (50°C) pipetira se po 5,5 ml inokulirane agarne podloge. Ploča se odmah stavi na nivelisanu podlogu, pokrije poklopcem i ohladi. Na takvu ploču stavljaju se metalni cilindri pomoću pincete, uz prikladnu šemu.
Pripremanje standardnih rastvora - standardni niz
Za izradu baždarnog dijagrama pomoću koga se izračunava koncentracija antibiotika u uzorku služi takozvani standardni niz - standardni rastvori četiri različite koncentracije, od kojih je jedan referentna koncentracija standarda. Standardni niz, odnosno četiri koncentracije standarda dobijaju se razređivanjem radnog rastvora standarda pomoću odgovarajućeg pufera ili posebno pripremljenog razređivača.
Pripremanje uzoraka za analizu
Uzorak za analizu priprema se zavisno od vrste uzorka, kao i vrste i koncentracije antibiotika. Potrebno je voditi računa o tome da ekstrakt uzorka bude razređen na osnovu označene ili pretpostavljene koncentracije antibiotika u izvornom uzorku u odnosu na koncentraciju koja je jednaka ili slična referentnoj koncentraciji standardnog niza.
Postupak određivanja
Svaka koncentracija standardnog niza stavi se na svaku od ukupno 10 Petrijevih šolja sa inokuliranom agarnom podlogom, i to u metalne cilindre raspoređene u obliku krsta. Rastvor uzorka stavi se u dva cilindra koji leže jedan nasuprot drugome, na svaku od pet Petrijevih ploča, dok se u preostala dva cilindra istih šolja stavi referentna koncentracija standardnog niza. Posle inkubacije inokulirane agarne podloge (ploče se ostave u termostatu preko noći na temperaturi 30°C) dobije se 10 zona inhibicija za rastvor uzorka i 10 zona inhibicija za referentnu koncentraciju na istim pločama. Takođe se dobije 10 zona inhibicije za svaku od četiri koncentracije standardnog niza (10 ploča).
Baždarni dijagram i izračunavanje
Posle inkubacije izmere se prečnici zona inhibicija dobijenih s pojedinim rastvorima standardnog niza i s rastvorom uzorka, pa se izračunaju njihove srednje vrednosti.
A |
- |
srednja vrednost svih prečnika zona inhibicije dobijenih s najvećom koncentracijom u standardnom nizu; |
B |
- |
srednja vrednost svih prečnika zona inhibicije dobijenih s referentnom koncentracijom standarda na pločama s uzorkom; |
C |
- |
srednja vrednost svih prečnika zona inhibicije dobijenih s onom koncentracijom iz standardnog niza koja je dobijena razređivanjem najveće koncentracije u standardnom nizu u odnosu 1 + 3; |
D |
- |
srednja vrednost svih prečnika zona inhibicija dobijenih s najmanjom koncentracijom u standardnom nizu (takva koncentracija dobijena je razređivanjem najveće koncentracije standardnog niza u odnosu 1 + 7. |
Baždarni dijagram se radi pomoću dve brojčane vrednosti (dobijene iz eksperimentalnih podataka), i to:
H = |
7A + 4B + C - 2D |
|
10 |
gde je:
H - korigovana srednja vrednost prečnika zona inhibicija dobijenih s najvećom koncentracijom u standardnom nizu:
L = |
7D + 4C + B - 2A |
|
10 |
gde je:
L - korigovana srednja vrednost prečnika zona inhibicija dobijenih s najmanjom koncentracijom u standardnom nizu.
Na apscisu (linerana skala) polulogaritamskog papira nanesu se korigovane srednje vrednosti H i L, a na ordinatu (logaritamska skala) - koncentracije standarda koje pripadaju tim rastvorima. Kroz prečnik linija povuče se prava linija.
Aktivnost antibiotika u rastvoru uzorka jednaka je vrednosti ordinate one tačke na baždarnom pravcu čija je apscisa jednaka korigovanoj srednjoj vrednosti prečnika zona inhibicija dobijenih rastvorom uzorka:
R korigovano = R + (M - N)
gde je:
Rkorigovano - korigovana srednja vrednost prečnika zona inhibicija dobijenih rastvorom uzorka;
M - korigovana srednja vrednost prečnika zona inhibicije dobijenih s referentnom koncentracijom;
R - srednja vrednost svih prečnika zona inhibicija dobijenih s rastvorom uzorka (prema pretpostavci, mora sadržati istu količinu antibiotika kao i referentna koncentracija u standardnom nizu);
N - srednja vrednost svih prečniha zona inhibicija dobijenih s referentnom koncentracijom u standardnom nizu, na pločama na kojima se ona nalazi zajedno s rastvorom uzorka.
45. Određivanje oksitetraciklin-hidrohlorida (OTC HCl)
A) Stočna hrana koja sadrži više od 50 mg/kg OTC HCl
Reagensi i test-mikroorganizam:
1) fosfatni pufer pH = 4,5 (u daljem tekstu: pufer);
2) rastvor hlorovodonične kiseline, c(HCl) = 0,1 mol/l,
3) rastvor natrijum-hidroksida, c(NaOH) = 1 mol/l;
4) hranljiva agarna podloga, pH = 5,6 - 5,7;
5) test-mikroorganizam: Bacillus cereus var. mycoides ATCC 9634 NCTC 10 380;
6) standard OTC HCl: kao standardni rastvor služi baza OTC, empirijske formule C22H24N2O9, teoretske aktivnosti 1000 µg/mg, koji se topi u 0,1 mol/HCl/l.
Postupak
Na analitičkoj vagi odmeri se oko 5,0 g homogenizovanog uzorka, kvantitativno prenese u tikvicu za jodni broj, zapremine 250 ml i doda tačno 100 ml 0,1 mol (HCl)/l. Sadržina tikvice mućka se na mehaničkoj mućkalici 1 h. Ekstrakt se profiltrira preko suvog, nabranog filtrir-papira. Alikvotni deo filtrata razređuje se puferom do koncentracije OTC od 0,4 µg/1 ml, što odgovara referentnoj koncentraciji standarda. Filtrat se razređuje na osnovu pretpostavljene količine OTC u uzorku.
Standard
U odmernu tikvicu zapremine 50 ml odmeri se odgovarajuća količina OTC baze, otopi u 0,1 mol (HCl)/l i istim rastvorom dopuni do oznake. To je radni rastvor standarda, koji se može upotrebljavati mesec dana ako se čuva na temperaturi + 4°C. Svaki mililitar radnog rastvora standarda mora sadržati 1000 µg OTC baze. Daljim razređivanjima puferom pripremi se standardni niz, i to:
0,8 µg/ml;
0,4 µg/ml - referentna koncentracija;
0,2 µg/ml;
0,1 µg/ml;
Rastvori uzorka i standardnog niza pripremljeni na taj način stavljaju se na Petrijeve ploče koje se inkubiraju prema opisanom postupku. Koncentracija OTC izračunava se prema opisanom postupku. S obzirom na to da je koncentracija antibiotika OTC izražena kao OTC baza, dobijenu vrednost treba pomnožiti faktorom 1,078 (odnos teoretske aktivnosti OTC baze i OTC HCl). Tada je koncentracija izražena u obliku OTC HCl.
B) Stočna hrana koja sadrži manje od 50 mg/kg OTC HCl
Za određivanje malih količina OTC u stočnoj hrani, uz ostale reagense opisane u prethodnoj metodi, upotrebljava se i razređivač.
Pripremanje uzorka
Na analitičkoj vagi odmeri se oko 10,0 g homogenizovanog uzorka, kvantitativno prenese u tikvicu za jodni broj, zapremine 250 ml i doda tačno 100 ml 0,1 mol (HCl)/l. Sadržina tikvice mućka se u mehaničkoj mućkalici 1 h i ekstrakt filtrira preko suvog, nabranog filtrir-papira. Tačno 20 ml ekstrakta otpipetira se u čašu zapremine 100 ml i pH vrednost rastvora podesi sa 1 mol (NaOH)/l na pH = 4,5. Zabeleži se utrošak baze i rastvoru doda toliko pufera da ukupna zapremina tečnosti u čaši bude 25 ml. Alikvotni deo ektrakta razredi se puferom na koncentraciju od 0,4 µg/ml (razređuje se na osnovu pretpostavljene količine OTC u stočnoj hrani). Koncentracija od 0,2 µg/ml, što odgovara referentnoj koncentraciji u standardnom nizu, dobija se razređivanjem razređivačem.
Pripremanje razređivača
Odmeri se ista količina slepe probe (bez OTC) i uzorka koji se analizira i podvrgne istom postupku ekstrakcije i korigovanja pH vrednosti. Tako pripremljen ekstrakt slepe probe razredi se puferom u istom odnosu u kome je razređen analogno pripremljen ekstrakt originalnog uzorka prilikom razređivanja na koncentraciju od 0,4 µg/ml.
Standard
Na analitičkoj vagi odmeri se 10,0 g slepe probe u tikvici za jodni broj i doda toliko OTC baze da koncentracija bude jednaka pretpostvljenoj količini OTC u uzorku. Dalji postupak je isti kao i pri pripremanju uzorka. Najveća koncentracija u standardnom nizu, u ovom slučaju 0,4 µg/ml, razređuje se razređivačem na koncentracije:
0,2 µg/ml - referentna koncentracija;
0,1 µg/ml;
0,05 µg/ml.
Rastvori uzorka i standarda pripremljeni na taj način stavljaju se na Petrijeve ploče, koje se inkubiraju prema opisanom postupku. Koncentracija OTC izračunava se takođe prema opisanom postupku.
46. Određivanje hlortetraciklin-hidrohlorida (CHTC HCl)
A) Stočna hrana koja sadrži više od 50 mg/kg CHTC HCl
Reagensi i test-mikroorganizam:
1) fosfatni pufer pH = 4,5 (u daljem tekstu: pufer);
2) aceton, p.a.;
3) koncentrovana hlorovodonična kiselina, HCl;
4) rastvor natrijum-hidroksida, c(NaOH) = 1 mol/l;
5) hranljiva agarna podloga, pH = 5,6 - 5,7;
6) test-mikroorganizam: Bacillus cereus var. mycoides ATCC 9633 NCTC 10380;
7) standard CHTC HCl: kao standard služi hlortetraciklin-hidrohlorid, empirijske formule C22H23N2O8Cl HCl, teoretske aktivnosti 1000 µg/ml, koji se topi u destilovanoj vodi.
Postupak
Na analitičkoj vagi odmeri se oko 5,0 g homogenizovanog uzorka, kvantitativno prenese u odmernu tikvicu zapremine 250 ml i doda tačno 100 ml kiselog rastvora acetona (650 ml acetona + 20 ml koncentrovane HCl + destilovane vode do 1000 ml). Sadržina tikvice mućka se 1 h na mehaničkoj mućkalici, a zatim rastvor dopuni do oznake fosfatnim puferom, izmeša se i profiltrira preko suvog, nabranog filtrir-papira. Alikvotni deo filtrata razređuje se puferom na koncentraciju CHTC HCl od 0,04 µg/ml, što odgovara referentnoj koncentraciji istog antibiotika u standardnom nizu. Alikvotni deo filtrata razređuje se na osnovu pretpostavljene količine CHTC HCl u uzorku.
Standard
U odmernu tikvicu zapremine 50 ml odmeri se odgovarajuća količina CHTC HCl standarda i rastvori u puferu, kojim se dopuni do oznake. To je radni rastvor standarda, koji u 1 ml mora sadržati 1000 µg CHTC HCl. Daljim razređivanjem radnog rastvora standarda puferom priprema se standardni niz, i to:
0,08 µg/ml;
0,04 µg/ml - referentna koncentracija;
0,02 µg/ml;
0,01 µg/ml.
Rastvori uzorka i standarda pripremljeni na taj način stavljaju se na Petrijeve ploče, koje se inkubiraju prema opisanom postupku. Koncentracije CHTC HCl izračuravaju se takođe prema opisanom postupku.
B) Stočna hrana koja sadrži manje od 50 mg/kg CHTC HCl
Za određivanje malih količina oleandomicina u stočnoj hrani, uz ostale reagense opisane u prethodnoj metodi, upotrebljava se i razređivač.
Pripremanje uzorka
Na analitičkoj vagi odmeri se oko 10,0 g homogenizovanog uzorka, kvantitativno prenese u tikvicu za jodni broj zapremine 250 ml, i doda tačno 100 ml kiselog rastvora acetona (650 ml acetona, 20 ml koncentrovane HCl i destilovane vode do 1000 ml). Sadržina tikvice mućka se 1 h na mehaničkoj mućkalici a ekstrakt se filtrira preko suvog, nabranog filtrir-papira. Tačno 20 ml ekstrakta otpipetira se u čašu zapremine 100 ml i pH vrednost rastvora podesi na 4,5 pomoću 1 mol (NaOH)/l. Zabeleži se utrošak baze i rastvoru se doda toliko pufera da ukupna zapremina tečnosti u čaši bude 25 ml. Rastvor se filtrira i alikvotni deo razredi puferom na koncentraciju od 0,08 µg/ml (na osnovu pretpostavljene količine navedenog antibiotika u uzorku). Taj rastvor se dalje razredi razređivačem na koncentraciju CHTC HCl od 0,04 µg/ml, što odgovara referentnoj koncentraciji standardnog niza.
Pripremanje razređivača
Odmeri se ista količina slepe probe (bez antibiotika) i uzorka koji se analizira i podvrgne istom postupku ekstrakcije i korigovanja pH vrednosti. Tako pripremljen ekstrakt slepe probe razredi sa fosfatnim puferom u istom odnosu u kome je razređen analogno pripremljen ekstrakt originalnog uzorka prilikom razređivanja na koncentraciju od 0,08 µg/ml.
Standard
Odmeri se 10,0 g slepe probe (stočna hrana bez prisustva antibiotika) u tikvicu za jodni broj i doda toliko CHTC HCl standarda da koncentracija odgovara pretpostavljenoj količini CHTC HCl u uzorku. Dalji postupak je isti kao i pri pripremanju uzorka (ekstrakcije, podešavanje pH vrednosti itd). Rastvor koncentracije 0,08 µg CHTC HCl/ml (najveća koncentracija u standardnom nizu) razređuje se razređivačem na koncentracije:
0,04 µg/ml - referentna koncentracija;
0,02 µg/ml;
0,01 µg/ml.
Rastvori uzorka i standarda pripremljeni na taj način stavljaju se na Petrijeve ploče, koje se inkubiraju prema opisanom postupku. Koncentracija CHTC HCl u izvorima izračunava se takođe prema opisanom postupku.
A) Stočna hrana koja sadrži više od 100 mg/kg oleandomicina
Reagensi i test-mikroorganizam:
1) fosfatno-bikarbonatni pufer, pH = 8 (u daljem tekstu pufer);
2) hranljiva agarna podloga, pH = 7,8 - 8,0;
3) metanol p.a.;
4) test-mikroorganizam:Sarcina lutea ATCC 9341;
5) standard oleandomicina: kao standard služi oleandomicin, empirijske formule C35H61NO12, teoretske aktivnosti 1000 µg/ml, topiv u metanolu.
Pripremanje uzorka
Na analitičkoj vagi odmeri se oko 5,0 g homogenizovanog uzorka, kvantitativno prenese u odmernu tikvicu zapremine 250 ml, doda se 150 ml pufera i 1 h mućka na mehaničkoj mućkalici .Rastvor se do oznake dopuni istim puferom, promeša i ekstrakt filtrira preko suvog, nabranog filtrir-papira. Alikvotni deo filtrata razređuje se puferom na koncentraciju 0,1 µg/ml, što odgovara referentnoj koncentraciji standarda, odnosno standardnog niza. Alikvotni deo filtrata razređuje se na osnovu pretpostavljene količine oleandomicina u uzorku.
Standard
U odmernu tikvicu zapremine 50 ml odmeri se odgovarajuća količina oleandomicin-standarda, rastvori u malo metanola i dobijeni rastvor razredi puferom i do oznake dopuni. To je radni standardni rastvor koji mora sadržati 1000 µg/ml oleandomicina. Daljim razređivanjima puferom pripremi se standardni niz, i to:
0,20 µg/ml;
0,10 µg/ml - referentna koncentracija;
0,05 µg/ml;
0,025 µg/ml.
Rastvori uzorka i standardnog niza pripremljeni na taj način stavljaju se na Petrijeve ploče, koje se inkubiraju prema opisanom postupku. Koncentracija oleandomicina izračunava se takođe prema opisanom postupku.
B) Stočna hrana koja sadrži manje od 100 mg/kg oleandomicina
Za određivanje malih količina oleandomicina u stočnoj hrani, uz ostale reagense i standarde opisane u prethodnoj metodi, upotrebljava se i razređivač.
Pripremanje uzorka
Na analitičkoj vagi se odmeri oko 10,0 g homogenizovanog uzorka, kvantitativno prenese u tikvicu za jodni broj zapremine 250 ml i doda tačno 100 ml pufera. Sadržina tikvice mućka se 1 h na mehaničkoj mućkalici, a ekstrakt filtrira preko suvog, nabranog filtrir-papira. Alikvotni deo ekstrakta razredi se istim puferom na koncentraciju 0,20 µg oleandomicina/1 ml (razređuje se na osnovu pretpostavljene količine oleandomicina u uzorku). Taj rastvor se razredi razređivačem na koncentraciju od 0,1 µg oleandomicina/1 ml, što odgovara referentnoj koncentraciji standardnog niza.
Pripremanje razređivača
Na analitičkoj vagi odmeri se oko 10 g slepe probe (bez antibiotika) i prebaci u tikvicu za jodni broj, zapremine 250 ml, i podvrgne istom postupku kao i analizirani uzorak. Tako pripremljen ekstrakt slepe probe razredi se puferom u istom odnosu u kome je razređen analogno pripremljen ekstrakt uzorka prilikom razređivanja na koncentraciju 0,20 µg oleandomicina/1 ml.
Standard
Na analitičkoj vagi odmeri se 10,0 g slepe probe u tikvicu za jodni broj zapremine 250 ml i doda toliko standarda oleandomicina da koncentracija bude jednaka pretpostavljenoj količini oleandomicina u uzorku. Dalji postupak je isti kao i za pripremanje uzorka (ekstrakcija, filtriranje itd.). Koncentracija od 0,20 µg oleandomicina u 1 ml (najveća koncentracija oleandomicina u standardnom nizu) razređuje se razređivačem na koncentracije:
0,1 µg/1 ml - referentna koncentracija;
0,05 µg/1 ml;
0,025 µg/1 ml.
Rastvori uzorka i standarda pripremljeni na taj način stavljaju se na Petrijeve ploče, koje se inkubiraju prema opisanom postupku. Koncentracija oleandomicina izračunava se takođe prema opisanom postupku.
48. Određivanje benzatin-penicilina G
A) Stočna hrana koja sadrži više od 100 mg/kg benzatin-penicilina G
Reagensi i test-mikroorganizam:
1) fosfatni pufer pH = 6 (u daljem tekstu: pufer);
2) N,N´ - dimetil-formamid, p.a.;
3) fiziološki rastvor;
4) hranljiva podloga, pH = 6,5 - 6,6;
5) test-mikroorganizam: Sarcina lutea ATCC 9341;
6) standard benzatin-penicilina G: kao standard služi benzatin-penicilin G, empirijske formule (C16H18N2O4S)2 · C16H20N2 · 4H2O, teoretske aktivnosti 1211 I. J./I mg, topi se u N,N´- dimetil-formamidu.
Postupak
Na analitičkoj vagi odmeri se oko 5,0 g homogenizovanog uzorka, kvantitativno prenese u odmernu tikvicu zapremine 250 ml, doda se 100 ml N,N´dimetil-formamida i sadržina tikvice mućka 1 h na mehaničkoj mućkalici. Volumen rastvora se dopuni zatim puferom do oznake, promeša i ekstrakt filtrira preko suvog, nabranog filtrir-papira. Alikvotni deo ekstrakta razređuje se puferom na koncentraciju 0,4 µg benzatin-penicilina G/1 ml, što odgovara referentnoj koncentraciji istog antibiotika u standardnom nizu. Razređenje se izvodi na osnovu pretpostavljene količine benzatin-penicilina G u uzorku.
Standard
U odmernu tikvicu zapremine 50 ml odmeri se odgovarajuća količina benzatin-penicilina G standarda, rastvori u N,N´-dimetil-formamidu i zapremina dopuni istim ratvorom do oznake. To je radni rastvor standarda koji u 1 ml mora sadržati 1000 µg benzatin-penicilina. Daljim razređivanjima puferom pripremi se standardni niz, i to:
0,8 µg/ml;
0,4 µg/ml - referentna koncentracija;
0,2 µg/ml;
0,1 µg/ml.
Rastvori uzorka i standardnog niza pripremljeni na taj način stavljaju se na Petrijeve ploče, koje se inkubiraju prema opisanom postpuku. Koncentracija benzatin-penicilina G izračunava se takođe prema opisanom postupku.
B) Stočna hrana koja sadrži manje od 100 mg/kg benzatin-penicilina G
Za određivanje malih količina benzatin-penicilina G u stočnoj hrani, uz ostale reagense opisane u prethodnoj metodi, upotrebljava se i razređivač.
Pripremanje uzorka
Na analitičkoj vagi odmeri se oko 10,0 g homogenizovanog uzorka, kvantitativno prenese u tikvicu za jodni broj, zapremine 250 ml, i doda 100 N,N´-dimetil-formamida. Sadržina tikvice mućka se 1 h na mehaničkoj mućkalici, a ekstrakt filtrira preko suvog, nabranog filtrir-papira. Alikvotni deo ekstrakta razredi se puferom na koncentraciju 0,4 µg/ml (na osnovu pretopostavljene količine navedenog antibiotika u uzorku). Taj rastvor se razredi razređivačem na koncentraciju 0,2 µg/ml, što odgovara referentnoj koncentraciji standardnog niza.
Pripremanje razređivača
Odmeri se ista količina slepe probe (bez antibiotika) i uzorka koji se analizira i podvrgne istom postupku ekstrakcije kao i analizirani uzorak. Tako pripremljen ekstrakt slepe probe razredi se puferom u istom odnosu u kome je razrađen analogno pripremljen ekstrakt uzorka prilikom razređivanja na koncentraciju 0,4 µg/ml benzatin-penicilina G.
Standard
Odmeri se 10,0 g slepe probe u tikvicu za jodni broj, zapremine 250 ml, i doda toliko benzatin-penicilina G standarda da koncentracija bude jednaka pretpostavljenoj količini benzatin-penicilina G u uzorku. Dalji postupak isti je kao i pri pripremanju uzorka. Koncentracija od 0,4 µg benzatin-penicilina G/1 ml (najveća koncentracija u standardnom nizu) razređuje se razređivačem na koncentracije:
0,20 µg/1 ml - referentna koncentracija;
0,10 µg/1 ml;
0,05 µg/1 ml.
Rastvori uzorka i standardnog niza pripremljeni na taj način stavljaju se na Petrijeve ploče, koje se inkubiraju prema opisanom postupku. Koncentracija benzatin-penicilina G izračunava se takođe prema opisanom postupku.
49. Određivanje cink-bacitracina
A) Stočna hrana koja sadrži više od 50 mg/kg cink-bacitracina
Reagensi i test-mikroorganizam:
1) fosfatni pufer pH = 6 (u daljem tekstu pufer);
2) hranljiva agarna podloga, pH = 6,5 - 6,6;
3) hlorovodonična kiselina, koncentrovana;
4) fiziološki rastvor za ispiranje test-mikroorganizma;
5) test-mikroorganizam Microccocus flavus ATCC 10240-A NCTC ili Sarcina lutea ATCC 9341;
6) standard cink-bacitracina: kao standard služi cink-bacitracin, empirijske formule C66H103O16N17C (1 molekul sadrži 1-2 ekvivalenta cinka). Teoretska aktivnost cink-bacitracina je 56 I.J./1 mg. Standard se topi u zakiseljenim vodenim rastvorima, u kojima je pH vrednost manja od 5.
Postupak
Na analitičkoj vagi odmeri se oko 5,0 g homogenizovanog uzorka, kvanititativno prenese u tikvicu za jodni broj zapremine 20 ml i doda tačno 25 ml razređene hlorovodonične kiseline (1 zapreminski deo koncentrovane HCl + 2,5 zapreminskih delova destilovane vode) i sadržina mućka 1 h na mehaničkoj mućkalici. Posle toga ekstraktu se doda 75 ml pufera i promeša. Posle filtriranja preko suvog, nabranog, filtrir-papira alikvotni deo ekstrakta se razredi na 0,1 µg cink-bacitracina/ /1 ml, što odgovara referentnoj koncentraciji u standardnom nizu. Alikvotni deo ekstrakta se razređuje na osnovu pretpostavljene količine cink-bacitracina u uzorku.
Standard
U odmernu tikvicu zapremine 50 ml odmeri se potrebna količina standarda cink-bacitracina, rastvori se u razređenoj hlorovodoničnoj kiselini (1 zapreminski deo koncentrovane HCl + 2,5 zapreminskih delova destilovane vode) i puferom dopuni do oznake. To je radni rastvor standarda, koji mora sadržati 1000 µg cink-bacitracina /1 ml. Daljim razređivanjima puferom pripremi se standardni niz, i to:
0,20 µg/1 ml;
0,10 µg/1 ml - referentna koncentracija;
0,05 µg/1 ml;
0,25 µg/1 ml.
Rastvori uzorka i standarda pripremljeni na taj način stavljaju se na Petrijeve ploče, koje se inkubiraju prema opisanom postupku. Koncentracija cink-bacitracina izračunava se takođe prema opisanom postupku.
B) Stočna hrana koja sadrži manje od 50 mg/kg cink-bacitracina
Za određivanje malih količina cink-bacitracina u stočnoj hrani, uz ostale reagense opisane u prethodnoj metodi, upotrebljava se i razređivač.
Pripremanje uzoraka
Na analitičkoj vagi odmeri se oko 10,0 g homogenizovanog uzorka, kvantitativno prenese u tikvicu za jodni broj, zapremine 250 ml i doda 25 ml razređene HCl (1 zapreminski deo koncentrovane HCl + 2,5 zapreminskih delova destilovane vode). Sadržina tikvice mućka se 1 h na mehaničkoj mućkalici. Posle toga, ekstraktu se doda tačno 75 ml pufera i promeša. Posle filtriranja ekstrakta preko suvog, nabranog filtrir-papira, alikvotni deo ekstrakta se razredi na koncentaciju cink-bacitracina od 0,2 µg/1 ml (na osnovu pretpostavljene količine cink-bacitracina u uzorku). Taj se rastvor razredi pomoću razređivača na koncentraciju od 0,1 µg cink-bacitracina/1 ml, što odgovara njegovoj referentnoj koncentraciji u standardnom nizu.
Pripremanje razređivača
Odmeri se oko 10,0 g slepe probe (bez cink-bacitracina) u tikvicu za jodni broj, zapremine 250 ml i podvrgne istom postupku kao i analizirani uzorak. Tako pripremljen ekstrakt slepe probe razredi se puferom u istom odnosu u kome je razređen analogno pripremljen ekstrakt uzorka prilikom razređivanja na koncentraciju 0,2 µg cink-bacitracina/1 ml.
Standard
Odmeri se 10,0 g slepe probe (bez antibiotika) u tikvicu za jodni broj, zapremine 250 ml, i doda toliko standarda cink-bacitracina da koncentracija bude jednaka pretpostavljenoj količini cink-bacitracina u uzorku. Dalji postupak je isti kao i pri pripremanju uzorka. Koncentracija od 0,20 µg/1 ml (najveća koncentracija u standardnom nizu) razređuje se razređivačem na koncentracije:
0,10 µg/1 ml - referentna koncentracija;
0,05 µg/1 ml;
0,025 µg/1 ml.
Rastvori uzorka i standarda pripremljeni na taj način stavljaju se na Petrijeve ploče, koje se inkubiraju prema opisanom postupku. Koncentracija cink-bacitracina izračunava se takođe prema opisanom postupku.
A) Stočna hrana koja sadrži više od 100 mg/kg tilozina
Reagensi i test mikroorganizam:
1) fosfatni pufer, pH = 8 (u daljem tekstu: pufer);
2) hranljiva agarna podloga, pH = 7,8 - 8,0;
3) metanol, p.a.;
4) test-mikroorganizam: Sarcina lutea ATCC 9341;
5) standard tilozina: kao standard služi tilozin-baza, teoretske aktivnosti 1050 µg/mg, koja se topi u metanolu.
Postupak
Na analitičkoj vagi se odmeri oko 5,0 g homogenizovanog uzorka, prenese kvantitativno u odmernu tikvicu zapremine 250 ml, doda 25 ml pufera i 75 ml mešavine metanola i pufera u odnosu 8 : 7. Sadržina tikvice mućka se 1 h, na mehaničkoj mućkalici, a zapremina u tikvici dopuni do oznake mešavinom metanola i pufera u odnosu 4 : 6. Dobijeni ekstrakt filtrira se kroz suvi, nabrani filtrir-papir i razređuje mešavinom metanola i pufera (4 + 6) na koncentraciju 0,50 µg tilozina/1 ml (razređuje se na osnovu pretpostavljene količine tilozina u uzorku). Koncentracija od 0,50 µg tilozina/1 ml odgovara referentnoj koncentraciji iz standardnog niza.
Standard
U odmernu tikvicu zapremine 50 ml odmeri se odgovarajuća količina standarda tilozina, rastvori se u malo metanola i dobijeni rastvor razredi do oznake mešavinom metanola i pufera (4 + 6). To je radni rastvor standarda, koji u 1 ml mora sadržati 1000 µg tilozina. Daljim razređivanjima istom mešavinom pripremi se standardni niz, i to:
1,00 µg/1 ml;
0,50 µg/1 ml - referentna koncentracija;
0,250 µg/1 ml;
0,125 µg/1 ml.
Rastvori uzorka i standarda pripremljeni na taj način stavljaju se na Petrijeve ploče, koje se inkubiraju prema opisanom postupku. Koncentracija tilozina izračunava se takođe prema priloženom postupku.
B) Stočna hrana koja sadrži manje od 100 mg/kg tilozina
Za određivanje malih količina tilozina u stočnoj hrani, uz ostale reagense opisane u prethodnoj metodi, upotrebljava se i razređivač.
Pripremanje uzorka
Na analitičkoj vagi odmeri se oko 10,0 g homogenizovanog uzorka, kvantitativno prenese u tikvicu za jodni broj, zapremine 250 ml i doda 25 ml pufera i 75 ml mešavine metanola i pufera (8 + 7). Sadržina tikvice se mućka 1 h na mehaničkoj mućkalici, ekstrakt dekantuje u odmernu tikvicu zapremine 250 ml, a ostatak u tikvici ekstrahuje se još jedanput pola sata mešavinom metanola i pufera (4 + 6). Taj drugi ekstrakt dekantuje se u istu odmernu tikvicu, talog se dva do tri puta ispere istim rastvaračem, posle čega se zapremina dopuni do oznake. Dobijeni ekstrakt profiltrira se preko suvog, nabranog filtrir-papira i alikvotni deo razredi mešavinom rastvarača metanola i pufera (4 + 6), na koncentraciju tilozina od 1 µg/1 ml (razređuje se na osnovu pretpostavljene količine tilozina u uzorku). Taj rastvor se razredi razređivačem na koncentraciju od 0,50 µg/1 ml, što odgovara referentnoj koncentraciji u standardnom nizu.
Pripremanje razređivača
Odmeri se oko 10,0 g slepe probe (bez antibiotika) u tikvicu za jodni broj, zapremine 250 ml, i podvrgne istom postupku kao i analizirani uzorak. Tako pripremljen ekstrakt slepe probe razredi se mešavinom metanola i pufera (4 + 6), u istom odnosu u kome je razređen analogno pripremljen ekstrakt uzorka prilikom razređivanja na koncentraciju 1 µg tilozina/1 ml.
Standard
Odmeri se oko 10,0 g slepe probe u tikvicu za jodni broj, zapremine 250 ml, i doda toliko standarda tilozina da koncentracija bude jednaka pretpostavljenoj količini tilozina u uzorku. Dalji postupak isti je kao i pri pripremanju uzorka (ekstrakcija, filtriranje itd). Koncentracija od 1 µg tilozina/1 ml (najveća koncentracija u standardnom nizu) razređuje se razređivačem na koncentracije:
0,50 µg/1 ml- referentna koncentracija;
0,25 µg/1 ml;
0,125 µg/1 ml.
Rastvori uzorka i standarda pripremljeni na taj način stavljaju se na Petrijeve ploče i inkubiraju prema opisanom postupku. Koncentracija tilozina takođe se izračunava prema opisanom postupku.
A) Stočna hrana koja sadrži više od 100 mg/kg neomicina
Reagensi i test-mikroorganizam:
Za određivanje koriste se sledeći reagensi:
1) fosfatni pufer, pH = 8 (u daljem tekstu: pufer);
2) hranljiva agarna podloga, pH = 7,8 - 8,0;
3) fiziološki rastvor;
4) test-mikroorganizam: Micrococcus pyogenes var. aureus ATCC 6538-P,
5) standard-neomicina: kao standard služi neomicin, empirijske formule C23H46N6O18, teoretske aktivnosti 1000 µg/mg, koji se topi u destilovanoj vodi.
Postupak
Na analitičkoj vagi odmeri se oko 5,0 g homogenizovanog uzorka, kvantitativno prenese u odmernu tikvicu zapremine 250 ml, doda se 100 ml pufera i sadržina tikvice mućka 1 h na mehaničkoj mućkalici. Ekstrakt se profiltrira preko suvog, nabranog filtrir-papira. Alikvotni deo ekstrakta razredi se puferom na koncentraciju od 1 µg neomicina/1 ml (razređuje se na osnovu pretpostavljene količine neomicina u uzorku). Koncentracija od 1 µg neomicina na 1 ml odgovara referentnoj koncentraciji iz standardnog niza.
Standard
U odmernu tikvicu zapremine 50 ml odmeri se potrebna količina standarda neomicina, rastvori se u malo destilovane vode i dobijeni rastvor puferom razredi do oznake. To je radni rastvor standarda koji u 1 ml sadrži 1000 µg neomicina. Daljim razređivanjima istim puferom pripremi se standardni niz, i to:
2,00 µg/1 ml;
1,00 µg/1 ml - referentna koncentracija;
0,50 µg/1 ml;
0,25 µg/1 ml.
Rastvori uzorka i standardnog niza pripremljeni na taj način stavljaju se na Petrijeve ploče, koje se inkubiraju prema opisanom postupku. Koncentracija neomicina izračunava se takođe prema opisanom postupku.
B) Stočna hrana koja sadrži manje od 100 mg/kg neomicina
Za određivanje malih količina neomicina u stočnoj hrani, uz ostale ragense upotrebljava se i razređivač.
Pripremanje uzorka
Odmeri se oko 10,0 g homogenizovanog uzorka, kvantitativno prenese u tikvicu za jodni broj, zapremine 250 ml i doda 100 ml pufera. Sadržina tikvice mućka se 1 h na mehaničkoj mućkalici. Dobijeni ekstrakt profiltrira se preko suvog, nabranog filtrir-papira, alikvotni deo razredi se istim puferom na koncentraciju 2,0 µg neomicina/1 ml (razređuje se na bazi pretpostavljene količine neomicina u uzorku). Taj rastvor se razredi razređivačem na koncentraciju 1 g neomicina na 1 ml, što odgovara referentnoj koncentraciji standardnog niza.
Pripremanje razređivača
Odmeri se oko 10,0 g slepe probe (bez antibiotika) u tikvicu za jodni broj zapremine 250 ml i podvrgne istom postupku kao i analizirani uzorak. Tako pripremljen ekstrakt slepe probe razredi se puferom u istom odnosu u kome je razređen analogno pripremljen ekstrakt uzorka prilikom razređivanja na koncentraciju 2,00 µg baze-neomicina/1 ml.
Standard
Odmeri se 10,0 g slepe probe u tikvicu za jodni broj, zapremine 250 ml i doda toliko standarda neomicina da koncentracija bude jednaka pretpostavljenoj količini neomicina u uzorku. Dalji postupak je isti kao i pri pripremanju uzorka (ekstrakcija, filtriranje itd.). Koncentracija od 2,00 µg neomicina/1 ml (najveća koncentracija neomicina u standardnom nizu) razređuje se razređivačem na koncentracije:
1,00 µg/1 ml - referentna koncentracija;
0,50 µg/1 ml;
0,025 µg/1 ml.
Rastvori uzorka i standarda pripremljeni na taj način stavljaju se na Petrijeve ploče, koje se inkubiraju prema opisanom postupku. Koncentracija neomicina izračunava se takođe prema opisanom postupku.
52. Određivanje virginiamicina
A) Stočna hrana koja sadrži više od 50 mg/kg virginiamicina
Reagensi i test-mikroorganizam:
1) metanol, p.a.;
2) hranljiva agarna podloga, pH = 7,4;
3) test-mikroorganizam:Sarcina lutea ATCC 9341;
4) standard virginiamicina: kao standard služi radni standard virginiamicina, teoretske aktivnosti 1900 I.J./1 mg, koji se tope u metanolu.
Pripremanje uzorka
Na analitičkoj vagi odmeri se oko 5,0 g homogenizovanog uzorka, kvantitativno prenese u odmernu tikvicu zapremine 250 ml, doda se 60 ml metanola i mućka 1 h na mehaničkoj mućkalici. Zapremina rastvora dopuni se do oznake destilovanom vodom i ekstrakt filtrira preko nabranog, suvog filtrir-papira. Alikvotni deo filtrata razređuje se vodom na koncentraciju 2,0 µg virginiamicina/1 ml, što odgovara referentnoj koncentraciji u standardnom nizu. Alikvotni deo filtrata razređuje se na osnovu pretpostavljene količine virginiamicina u uzorku.
Standard
U odmernu tikvicu zapremine 50 ml odmeri se potrebna količina standarda virginiamicina, rastvori u malo metanola i dobijeni rastvor razredi do oznake destilovanom vodom. To je radni rastvor standarda, koji u 1 ml mora sadržati 1000 µg virginiamicina. Daljim razređivanjima destilovanom vodom pripremi se standardni niz, i to:
4,00 µg/1 ml;
2,00 µg/1 ml - referentna koncentracija;
1,00 µg/1 ml;
0,50 µg/1 ml.
Rastvori uzorka i standarda pripremljeni na taj način stavljaju se na Petrijeve ploče, koje se inkubiraju prema opisanom postupku. Koncentracija virginiamicina izračunava se takođe prema opisanom postupku.