
B.40. IN VITRO ISPITIVANJE KOROZIVNOG OŠTEĆENJA KOŽE: ISPITIVANJE TRANSKUTANOG ELEKTRIČNOG OTPORA (TER)
Ova metoda potpuno odgovara metodi OECD: TG 430 (2004).
Korozivno oštećenje kože odnosi se na stvaranje ireverzibilnog oštećenja tkiva u koži po nanošenju materijala koji se ispituje (kao što je definisano u GHS)1. Ova metoda obezbeđuje proceduru kojom se procena korozivnosti ne izvodi na živim životinjama.
Procena korozivnosti za kožu uključuje upotrebu laboratorijskih životinja2. Zabrinutost zbog bola i patnje životinja koje su uključene u ovaj postupak navodi se u reviziji metode V.4. koja je data u ovom prilogu, a koja dozvoljava određivanje korozivnog oštećenja kože upotrebom alternativnih, in vitro metoda, da bi se izbegli bol i patnja životinja.
Prvi korak ka definisanju alternativnih ispitivanja koji se mogu koristiti za ispitivanje korozivnosti za kožu u regulatorne svrhe bilo je izvođenje prevalidacionih studija3. Potom je izvedena6, 7, 8 formalna validacija studija in vitro metoda za procenu korozivnog oštećenja kože4, 5. Ishod ovih studija i druga objavljena literatura doveli su do preporuke da se sledeća ispitivanja mogu koristiti za procenu in vivo korozivnosti za kožu9, 10, 11: ispitivanje sa modelom humane kože (videti metodu V.40 bis koja je data u ovom prilogu) i ispitivanje transkutanog električnog otpora (ova metoda).
Studija validacije i druge objavljene studije potvrđuju da je ispitivanje transkutanog električnog otpora (TER) kože pacova12, 13 moguće pouzdano napraviti razliku između poznatih koroziva za kožu i nekoroziva5, 9.
Ispitivanje koje se opisuje u ovoj metodi omogućava identifikaciju korozivnih hemijskih supstanci i smeša. To dalje omogućava identifikaciju supstanci i smeša koje nisu korozivne kada je to potkrepljeno određivanjem težine dokaza korišćenjem (na osnovu) postojećih informacija (npr. pH, odnos struktura-aktivnost, humani ili životinjski podaci)1, 2, 11, 14. To ne daje podatak o iritaciji kože, niti dozvoljava subklasifikaciju korozivne supstance kao što je to dozvoljeno u GHS1.
Za potpunu procenu lokalnih efekata na koži posle jednokratnog izlaganja putem kože, preporučuje se da se prati sledeća strategija ispitivanja kao što je dodato u ispitivanju metodi V.4.2 koja je data u ovom prilogu i kao što je dato u GHS1. Ova strategija ispitivanja uključuje izvođenje in vitro ispitivanje za korozivno oštećenje kože (kao što je opisano u ovoj metodi) i iritacije kože pre razmatranja ispitivanja na živim životinjama.
Korozivno oštećenje in vivo jeste nastanak ireverzibilnih oštećenja kože odnosno vidljive nekroze u epidermisu i dermisu nakon četvoročasovne primene ispitivane supstance. Korozivno oštećenje kože karakteriše se prema tipu izazvanog oštećenja kao: ulkus, krvarenje ili krvave kraste i na kraju perioda posmatranja od 14 dana prema promeni boje kože uslovljene perutanjem i pojavi površina sa alopecijom i ožiljnog tkiva. U cilju procene nejasnih oštećenja obavljaju se histopatološka ispitivanja.
Transkutani električni otpor (u daljem tekstu: TER) jeste mera električne impedance kože, kao vrednost otpora u kilo omima. Jednostavna i snažna metoda procene funkcije barijere snimanjem prolaska jona kroz kožu pomoću aparature Wheatstone bridge.
Tabela 1. Referentne hemikalije
Naziv | EINECS broj | CAS broj |
|
1,2-diaminopropan | 201-155-9 | 78-90-0 | jako korozivno |
akrilna kiselina | 201-177-9 | 79-10-7 | jako korozivno |
2-terc- butilfenol | 201-807-2 | 88-18-6 | korozivno |
Kalijum-hidroksid (10 %) | 215-181-3 | 1310-58-3 | korozivno |
sumporna kiselina (10 %) | 231-639-5 | 7664-93-9 | korozivno |
oktanska kiselina (kaprilna kiselina) | 204-677-5 | 124-07-02 | korozivno |
4-amino-1,2,4-triazol | 209-533-5 | 584-13-4 | nije korozivno |
eugenol | 202-589-1 | 97-53-0 | nije korozivno |
Fenetil-bromid | 203-130-8 | 103-63-9 | nije korozivno |
tetrahloretilen | 204-825-9 | 27-18-4 | nije korozivno |
izostearinska kiselina | 250-178-0 | 30399-84-9 | nije korozivno |
4-(metiltio)-benzaldehid | 222-365-7 | 3446-89-7 | nije korozivno |
Većina navedenih hemikalija preuzeta je sa spiska hemikalija izabranih za međunarodnu studiju validacije ECVAM4. Njihov izbor se bazira na sledećim kriterijumima:
a) jednak broj korozivnih i nekorozivnih supstanci;
b) komercijalno dostupne supstance koje pokrivaju većinu relevantnih hemijskih klasa;
v) obuhvatanje jako korozivnih kao i manje korozivnih supstanci kako bi se omogućilo razlikovanje na osnovu korozivnog potencijala;
g) izbor hemikalija kojima se može rukovati u laboratoriji bez postavljanja drugih ozbiljnih opasnosti osim korozivnosti.
Hemikalija koja se ispituje primenjuje se do 24 sata na epidermalne površine diskova kože u sistemu za ispitivanje koji se sastoji od dve komore u kome diskovi kože funkcionišu kao pregrada između komora. Diskovi kože su uzeti od humano usmrćenih pacova starosti 28 do 30 dana. Korozivni materijali se identifikuju prema njihovom svojstvu da produkuju gubitak integriteta i zaštitne funkcije stratum corneum-a, što se meri kao smanjenje TER ispod graničnog nivoa12. Za TER pacova, granična vrednost od 5 kΩ izabrana je na osnovu opsežnih podataka za širok opseg hemikalija gde je velika većina vrednosti bila ili jasno dosta iznad (često >10 kΩ ili dosta ispod (često <3 kΩ) ove vrednosti12. Materijali koji nisu korozivni kod životinja ali jesu ili nisu iritabilni ne snižavaju TER ispod ove granične vrednosti. Korićenje drugih preparata kože ili druge opreme može da izmeni graničnu vrednost i zahteva dalju validaciju. Korak vezivanja boje uključen je u postupak ispitivanja za potvrdu pozitivnih rezultata TER uključujući vrednosti približne 5kΩ.
Korak vezivanja boje određuje da li je povećanje u propustljivosti jona posledica fizičkog oštećenja stratum corneum-a. Metoda ispitivanja TER koja koristi kožu pacova ukazuje na in vivo korozivnost na kunićima koja se procenjuje u metodi ispitivanja V.4. koja je data u ovom prilogu2. Treba napomenuti da je in vivo ispitivanje na kunićima visoko konzervativan u pogledu na korozivnost i iritabilnost za kožu u poređenju sa "patch" testom na humanoj koži15.
1.5.1. Životinje
Koriste se pacovi jer je osetljivost njihove kože na hemikalije u ovoj metodi prethodno prikazana10. Starost (u trenutku uzimanja kože) i soj pacova su veoma važni kako bi se obezbedilo da su folikuli dlake u fazi mirovanja pre nego što počne rast krzna odraslih.
Dlaka na leđima i boku mladih, približno 22 dana starih mužjaka ili ženki pacova (izvedenih od Wistar ili sličnog soja) pažljivo se ukloni pomoću malih makaza. Onda se životinje peru pažljivim brisanjem, dok se ograničeni deo potapa u rastvor antibiotika (koji sadrži, npr. streptomicin, penicilin, hloramfenikol i amfotericin, u koncentracijama koje imaju efekat inhibicije rasta bakterija). Životinje se peru antibioticima ponovo trećeg i četvrtog dana posle prvog pranja i koriste se tokom tri dana posle drugog pranja, kada se stratum corneum oporavio od odstranjivanja dlake.
1.5.2. Priprema diskova kože
Životinje se humano žrtvuju kada su 28 do 30 dana stari. Ovo doba je kritično. Dorzalno-lateralna koža svake životinje ukloni se i skine se prekomerna subkutana masnoća pažljivim guljenjem sa kože. Diskovi kože prečnika od oko 20 mm se uklone. Koža se može čuvati pre nego što se diskovi upotrebe kada je pokazano da su podaci pozitivne i negativne kontrole jednaki onima koji se dobiju sa svežom kožom.
Svaki disk kože se postavlja preko jednog od krajeva politetrafluoroetilenske (u daljem tekstu: PTFE) cevi, obezbeđujući da je epidermalna površina u kontaktu sa cevi. Gumeni "O" prsten stavlja se na kraj cevi kako bi držao kožu na mestu i višak tkiva se odstranjuje. Dimenzije cevi i "O" prstena prikazane su na Slici 2. Gumeni "O" prsten se tada pažljivo pečati za kraj PTFE cevi vazelinom. Cev se podupire opružnom podloškom unutar receptorske komore koja sadrži rastvor magnezijum sulfata (154 mM) (Slika 1). Disk kože treba da je potpuno uronjen u rastvor MgSO4. Čak 10 do 15 diskova kože može se dobiti od jedne kože pacova.
Pre nego što počne ispitivanje, meri se električni otpor dva diska kože kao postupak kontrole kvaliteta za svaku kožu životinje. Oba diska treba da daju vrednost otpora veću od 10 kΩ kako bi se preostali diskovi koristili u ispitivanju. Ukoliko je vrednost otpora manja od 10 kΩ, preostali diskovi te kože treba da budu odbačeni.
1.5.3. Primena ispitivanih i kontrolnih supstanci
Tečne ispitivane supstance (150 μL) ravnomerno se nanose na epidermalnu površinu unutar cevi. Prilikom ispitivanja čvrstih materijala, nanosi se dovoljna količina čvrste materije ujednačeno na disk tako da čitava površina epidermisa bude pokrivena. Preko čvrste materije dodaje se dejonizovana voda (150 μL) i cev se blago prodrma. Kako bi se postigao maksimalan kontakt s kožom, čvrste materije je možda potrebno zagrejati do temperature od 30°C da bi se otopile ili je potrebno omekšati ispitivanu supstancu ili je usitniti da se napravi zrnasti materijal ili prah.
Tri diska kože koriste se za svaku ispitivanu i kontrolnu supstancu. Ispitivane supstance se primenjuju 24 sata pri temperaturi od 20°C do 23°C. Ispitivana supstanca se uklanja pranjem pod mlazom vode temperature do 30°C sve dok se više supstance ne može odstraniti.
1.5.4. TER merenja
Impedanca kože se meri tako što se meri TER niskovoltažnim uređajem s naizmeničnom strujom Wheatstone databridge13. Opšte karakteristike mosta su radni napon 1-3 Volt-a, naizmenična struja od 50 Hz do 1.000 Hz sinusnog ili četvrtastog oblika i opseg merenja od najmanje 0,1 kΩ do 30 kΩ. "Databridge" koji je korišćen u studiji validacije gde je merio induktivnost, kapacitivnost i otpor do vrednosti od redom 2.000 H, 2.000 μF i 2 MΩ, pri frekvencijama od 100 Hz ili 1 kHz koristeći redne ili paralelne vrednosti. U svrhu TER korozivnosti, merenja u ispitivanju se snimaju u otporu, pri frekvenci od 100 Hz i korišćenjem rednih vrednosti. Pre merenja električnog otpora površinski napon kože se smanjuje dodavanjem dovoljne zapremine 70% etanola da pokrije epiderm. Posle nekoliko sekundi etanol se uklanja iz cevi i tkivo se tada hidrira dodavanjem rastvora 3 mL rastvora magnezijum sulfata (154 mM). "Databridge" elektrode se postavljaju na obe strane diskova kože kako bi se izmerio otpor u kΩ/disk kože (Slika 1). Dimenzije elektroda i dužina elektroda koja je izložena ispod štipaljki prikazane su na Slici 2. Štipaljka koja je zakačena na unutrašnju elektrodu otvorena je na vrhu PTFE cevi tokom merenja otpora kako bi se obezbedilo da je dužina elektrode koja je uronjena u rastvor MgSO4 stalna. Spoljna elektroda se nalazi unutar receptorske komore tako da naleže na dno komore. Razmak između opružne podloške i dna PTFE cevi održava se konstantnim (Slika 2), budući da ovaj razmak utiče na dobijene vrednosti otpora. Zbog toga, razmak između unutrašnje elektrode i diska kože treba da bude konstantan i minimalan (1 mm do 2 mm).
Ukoliko je izmerena vrednost otpora veća od 20kΩ, to može biti zbog toga što ostaci ispitivane supstance prekrivaju epidermalnu površinu diska kože. Može se pokušati sa daljim odstranjivanjem ovog sloja, npr. začepljivanjem PTFE cevi palcem u rukavici i protresanjem približno 10 sekundi. Rastvor magnezijum sulfata se odstranjuje i merenje otpora se ponavlja sa svežim magnezijum sulfatom.
Svojstva i dimenzije aparature za ispitivanje i postupka ispitivanja koji se koriste mogu da utiču na dobijene vrednosti TER. Granična vrednost od 5 kΩ je izvedena iz podataka dobijenih sa specifičnom aparaturom i postupkom koji su opisani u ovoj metodi. Drugačija granična vrednost i vrednosti kontrole mogu se primeniti ukoliko su uslovi ispitivanja izmenjeni ili se koristi drugačija aparatura. Zbog toga je neophodno kalibrisati ovu metodologiju i vrednosti graničnog otpora ispitivanjem serije referentnih standarda koji su izabrani od hemikalija korićenih u studiji validacije4, 5 ili od hemijskih klasa sličnih hemikaliji koja se ispituje. Grupa odgovarajućih referentnih hemikalija data je u Tabeli 1.
1.5.5. Metode vezivanja boje
Ekspozicija određenim materijalima koji nisu korozivni može da ima za posledicu smanjenje otpora ispod granice od 5 kΩ dozvoljavajući prolazak jona kroz stratum corneum tako smanjujući električni otpor5. Na primer, neutralne organske i hemikalije koje imaju površinski aktivne osobine (uključujući detergente, emulgatore i druge surfaktante) mogu da uklone lipide kože čineći barijeru više propustljivom za jone. Ako su TER vrednosti ispitivane supstance manje od ili približno 5 kΩ u odsustvu vidljivog oštećenja, procena prodiranja boje treba da se izvede na kontrolnim i tretiranim tkivima da bi se odredilo da li su dobijene TER vrednosti rezultat povećane propustljivosti kože ili korozivno oštećenje kože3, 5. U drugom slučaju kada postoji prekid u stratum corneum-u, boja sulforodamin B, kada se nanese na površinu kože brzo prodire i boji potporno tkivo. Ova određena boja je stabilna prema širokom opsegu hemikalija i na rezultate ne utiče proces ekstrakcije koji je opisan ispod.
1.5.5.1. Nanošenje i uklanjanje sulforodamin B boje
Posle procene TER, magnezijum sulfat se odstranjuje iz cevi i koža se pažljivo pregleda kako bi se uočilo oštećenje. Ukoliko nema vidljivog većeg oštećenja, boja sulforodamin B (Acid Red 52; C.I. 45100; EINECS broj 222-529-8; CAS broj 3520-42-1), 150 µL 10% (w/v) rastvor u destilovanoj vodi nanosi se na epidermalnu površinu svakog diska kože na dva sata. Diskovi kože se tada ispiraju sa vodom sa česme pri najviše sobnoj temperaturi približno 10 sekundi kako bi se odstranila sva prekomerna/nevezana boja. Svaki disk kože pažljivo se ukloni sa PTFE cevi i stavlja u bočicu (npr. 20 mL staklena scintilaciona bočica) koja sadrži dejonizovanu vodu (8 mL). Bočice se lagano mešaju pet minuta kako bi se odstranila dodatna nevezana boja. Ovaj postupak ispiranja se zatim ponavlja, posle čega se diskovi kože uklanjaju i stavljaju u bočice koje sadže 5mL 30% (w/v) natrijum dodecilsulfata (SDS) u destilovanoj vodi i inkubiraju se preko noći na 60°C.
Posle inkubacije svaki disk kože se odstranjuje i baca, a preostali rastvor se centrifugira osam minuta pri 21°C (relativna centrifugalna sila je oko 175 x g). Uzorak od 1 mL supernatanta se razblažuje 1 u 5 (v/v) (tj. 1 mL + 4 mL) sa 30% (w/v) SDS u destilovanoj vodi. Optička gustina (u daljem tekstu: OD) rastvora meri se na 565 nm.
1.5.5.2. Izračunavanje sadržaja boje
Sadržaj boje sulforodamin B po disku izračunava se iz OD vrednosti5 (koeficijent molarne estinkcije boje sulforodamin B pri 565 nm = 8,7 x 104; molekularna masa = 580). Sadržaj boje sulforodamin B određuje se za svaki disk kože korišćenjem odgovarajuće kalibracione krive i zatim se izračunava srednja vrednost sadržaja boje za replikate.
Vrednosti otpora (kΩ) i srednje vrednosti sadržaja boje(μg/disk), kada to odgovara, za ispitivani materijal, kao i za pozitivne i negativne kontrole treba da budu navedene tabelarno (pojedinačni podaci ispitivanja i srednje vrednosti ±S.D.), uključujući podatke za ponovljene eksperimente, srednje vrednosti i pojedinačne vrednosti.
Srednje vrednosti TER prihvataju se ako su vrednosti istovremene pozitivne i negativne kontrole u prihvatljivim okvirima za metodu u laboratoriji koja vrši ispitivanje. Prihvatljivi opsezi otpora za metodologiju i aparaturu koje su opisane dati su u sledećoj tabeli:
Tabela 1.
kontrola | supstanca | opseg otpora (kΩ) |
pozitivna | 10M hlorovodonična kiselina | 0,5 - 1,0 |
negativna | destilovana voda | 10 - 25 |
Rezultati srednje vrednosti sadržaja boje prihvataju se pod uslovom da se vrednosti istovremene kontrole nalaze u okviru prihvatljivih opsega za metodu. Predloženi prihvatljivi opsezi sadržaja boje za kontrolne supstance za metodologiju i aparaturu koje su opisani su:
Tabela 2.
kontrola | supstanca | opseg sadržaja boje (μg/disk) |
pozitivna | 10M hlorovodonična kiselina | 40 - 100 |
negativna | destilovana voda | 15 - 35 |
Supstanca koja se ispituje ne smatra se korozivnom za kožu:
a) ako je srednja vrednost TER dobijena za ispitivanu supstancu veća od 5 kΩ ili
b) ako je srednja vrednost TER manja ili jednaka 5 kΩ i
- disk kože ne pokazuje vidljivo oštećenje i
- srednja vrednost sadržaja boje diska je daleko ispod srednjeg sadržaja boje diska istovremene pozitivne kontrole, 10M HCl.
Ispitivana supstanca se smatra korozivnom za kožu:
a) ako je srednja vrednost TER manja ili jednaka 5 kΩ i disk kože je vidljivo oštećen ili
b) ako je srednja vrednost TER manja ili jednaka 5 kΩ i
- disk kože ne pokazuje vidljivo oštećenje, ali
- srednja vrednost sadržaja boje diska je veća ili jednaka srednjoj vrednosti sadržaja boje diska istovremene pozitivne kontrole, 10M HCl.
Izveštaj o ispitivanju sadrži podatke o:
1) Ispitivanim i kontrolnim supstancama:
- hemijsko ime kao što je IUPAC ili CAS ime i CAS broj, ako je poznato;
- čistoća i sastav supstance ili smeše (u težinskim procentima) i fizička priroda;
- fizičke i hemijske osobine kao što su fizičko stanje, pH, stabilnost, rastvorljivost u vodi, ono što je relevantno za izvođenje studije;
- tretman ispitivane/kontrolne supstance pre ispitivanja, ako je primenljivo (npr. zagrevanje, usitnjavanje);
- stabilnost, ako je poznata.
2) Eksperimentalnim životinjama:
- korišćen soj i pol;
- starost životinja u trenutku kada se koriste kao donorske životinje;
- izvor, uslovi smeštaja, ishrana, itd.;
- detalji pripremanja kože.
3) Uslovima ispitivanja:
- kalibracione krive za aparaturu na kojoj se vrši ispitivanje;
- kalibracione krive za izvođenje ispitivanja vezivanja boje;
- detalji postupka ispitivanja koji se koristi za merenja TER;
- detalji postupka ispitivanja koji se koristi za procenu vezivanja boje, ako odgovara;
- opis svake izmene postupka ispitivanja;
- opis korišćenih kriterijuma za ocenjivanje.
4) Rezultatima:
- u tabeli dati podaci ispitivanja TER i vezivanja boje (ako je odgovarajuće) za svaku pojedinačnu životinju i uzorak kože;
- opis svakog primećenog efekta.
5) Obrazloženju rezultata.
6) Zaključcima.
1. OECD, (2001) Harmonised Integrated Classification System for Human Health and Environmental Hazards of Chemical Substances and Mixtures. OECD Series on Testing and Assessment Number 33. ENV/JM/MONO(2001)6, Paris. http://www.olis.oecd.org/olis/2001doc.nsf/LinkTo/env-jm-mono(2001)6.
2. Testing Method B.4. Acute Toxicity: Dermal Irritation/Corrosion.
3. Botham, P.A., Chamberlain, M., Barratt, M.D., Curren, R.D., Esdaile, D.J., Gardner, J.R., Gordon, V.C., Hildebrand, B., Lewis, R.W., Liebsch, M., Logemann, P., Osborne, R., Ponec, M., Regnier, J.F., Steiling, W., Walker, A.P., and Balls, M., (1995) A prevalidation study on in vitro skin corrosivity testing. The report and recommendations of ECVAM Workshop 6. ATLA 23, p. 219-255.
4. Barratt, M.D., Brantom, P.G., Fentem, J.H., Gerner, I., Walker, A.P., and Worth, A.P. (1998). The ECVAM international validation study on in vitro tests for skin corrosivity. 1. Selection and distribution of the test chemicals. Toxicology. In vitro 12, p. 471-482.
5. Fentem, J.H., Archer, G.E.B., Balls, M., Botham, P.A., Curren, R.D., Earl, L.K., Esdaile, D.J., Holzhütter, H.- G., and Liebsch, M., (1998) The ECVAM international validation study on in vitro tests for skin corrosivity. 2. Results and evaluation by the Management Team. Toxicology. In vitro 12, p. 483- 524.
6. OECD, (1996) Final Report of the OECD Workshop on Harmonisation of Validation and Acceptance Criteria for Alternative Toxicological Test Methods, p. 62.
7. Balls, M., Blaauboer, B.J., Fentem. J.H., Bruner. L., Combes, R.D., Ekwall, B., Fielder. R.J., Guillouzo, A., Lewis, R.W., Lovell, D.P., Reinhardt, C.A., Repetto, G., Sladowski. D., Spielmann, H., and Zucco, F., (1995) Practical aspects of the validation of toxicity test procedures. The report and recommendations of ECVAM workshops. ATLA 23, p. 129-147.
8. ICCVAM, (Interagency Coordinating Committee on the Validation of Alternative Methods)., (1997) Validation and Regulatory Acceptance of Toxicological Test Methods. NIH Publication No 97-3981. National Institute of Environmental Health Sciences, Research Triangle Park, NC, USA. http://iccvam.niehs.nih.gov/docs/guidelines/validate.pdf.
9. ECVAM, (1998) ECVAM News & Views. ATLA 26, 275-280.
10. ICCVAM (Interagency Coordinating Committee on the Validation of Alternative Methods)., (2002) ICCVAM evaluation of EpiDermTM, EPISKINTM (EPI-200), and the Rat Skin Transcutaneous Electrical Resistance (TER) assay: In vitro test methods for assessing dermal corrosivity potential of chemicals. NIH Publication No 02-4502. National Toxicology Program Interagency Center for the Evaluation of Alternative Toxicological Methods, National Institute of Environmental Health Sciences, Research Triangle Park, NC, USA. http://iccvam.niehs.nih.gov/methods/epiddocs/epis_brd.pdf.
11. OECD (2002) Extended Expert Consultation Meeting on The In vitro Skin Corrosion Test Guideline Proposal, Berlin, 1st-2nd November 2001, Secretariat's Final Summary Report, 27th March 2002, OECD ENV/EHS, available upon request from the Secretariat.
12. Oliver, G.J.A., Pemberton, M.A., and Rhodes, C., (1986) An in vitro skin corrosivity test-modificationsand validation. Fd. Chem. Toxicol. 24, 507-512.
13. Botham, P.A., Hall, T.J., Dennett, R., McCall, J.C., Basketter, D.A., Whittle, E., Cheeseman, M., Esdaile, D.J., and Gardner, J., (1992) The skin corrosivity test in vitro: results of an interlaboratory trial. Toxic. In vitro 6, p. 191-194.
14. Worth A.P., Fentem J.H., Balls, M., Botham, P.A., Curren, R.D., Earl, L.K., Esdaile D.J., Liebsch, M., (1998) An Evaluation of the Proposed OECD Testing Strategy for Skin Corrosion. ATLA 26, p. 709-720
15. Basketter, D.A., Chamberlain, M., Griffiths, H.A., Rowson, M., Whittle, E., York, M., (1997) The classification of skin irritants by human patch test. Fd. Chem. Toxicol. 35, p. 845-852.
16. Oliver G.J.A, Pemberton M.A and Rhodes C., (1988) An In vitro model for identifying skin-corrosive chemicals. I. Initial Validation. Toxicology In vitro. 2, p. 7-17.
Slika 1
Aparatura za ispitivanje kože pacova TER ogledom
Slika 2: Dimenzije politetrafluoroetilenske (PFTE) i receptorske cevi i elektroda koje se koriste

Kritični faktori aparature koja je prikazana:
- unutrašnji promer PTFE cevi;
- dužina elektrode u odnosu na PTFE cev i receptorsku cev, tako da elektroda ne dotiče disk kože i da je standardna dužina elektrode u kontaktu sa rastvorom magnezijum sulfata;
- količina rastvora magnezijum sulfata u receptorskoj cevi treba da da dubinu tečnosti, u odnosu na nivo u PTFE cev, kao što je prikazano na Slici 1;
- disk kože treba da bude dovoljno dobro pričvršćen za PTFE cev, tako da električni otpor bude prava mera svojstava kože.
B.40. BIS. IN VITRO KOROZIVNO OŠTEĆENJE KOŽE: ISPITIVANJE NA MODELU HUMANE KOŽE
Ova metoda potpuno odgovara metodi OECD: TG 431 (2004).
Korozivno oštećenje kože odnosi se na stvaranje ireverzibilnog oštećenja tkiva u koži po nanošenju materijala koji se ispituje (kao što je definisano u GHS)1. Ova metoda ispitivanja ne zahteva upotrebu živih životinja ili životinjskog tkiva za procenu korozivnosti za kožu.
Procena korozivnosti za kožu uključuje upotrebu laboratorijskih životinja2. Zabrinutost zbog bola i patnje životinja koje su uključene u ovaj postupak pominje se u reviziji metode V.4. koja je data u ovom prilogu i koja dozvoljava određivanje korozivnog oštećenja kože upotrebom alternativnih, in vitro metoda, da bi se izbegli bol i patnja životinja.
Prvi korak ka definisanju alternativnih ispitivanja koji se mogu koristiti za ispitivanje korozivnosti za kožu u regulatorne svrhe bilo je izvođenje prevalidacionih studija3. Potom je izvedena6, 7, 8 formalna validacija studija in vitro metoda za procenu korozivnog oštećenja kože4, 5. Ishod ovih studija i druga objavljena literatura9 doveli su do preporuke da se sledeća ispitivanja mogu koristiti za procenu in vivo korozivnosti za kožu10, 11, 12, 13: ispitivanje sa modelom humane kože (ova metoda) i ispitivanje transkutanog električnog otpora (videti metodu V.40. koja je data u ovom prilogu).
Studije validacije potvrđuju da je ispitivanjima a koji uključuju modele humane kože3, 4, 5, 9 moguće pouzdano napraviti razliku između poznatih korozivnih i nekorozivnih supstanci za kožu. Postupak ispitivanja može da obezbedi naznaku razlike između jakih i manje jakih koroziva za kožu.
Ispitivanje koje se opisuje u ovoj metodi omogućava identifikaciju korozivnih hemijskih supstanci i smeša. To dalje omogućava identifikaciju supstanci i smeša koje nisu korozivne kada je to potkrepljeno određivanjem težine dokaza korišćenjem (na osnovu) postojećih podataka (npr. pH, odnos struktura-aktivnost, humani i/ili životinjski podaci)1, 2, 13, 14. To obično ne daje odgovarajuće podatke o iritaciji kože, niti dozvoljava subklasifikaciju korozivne supstance kao što je to dozvoljeno u GHS1.
Za potpunu procenu lokalnih efekata na koži posle jednokratnog izlaganja putem kože, preporučuje se da se prati sledeća strategija ispitivanja kao što je dato u metodi ispitivanja V.4. koja je data u ovom prilogu2 i kao što je dato u GHS1. Ova strategija ispitivanja uključuje izvođenje in vitro ispitivanja za korozivno oštećenje kože (kao što je opisano u ovoj metodi) i iritacije kože pre razmatranja ispitivanja na živim životinjama.
Korozivno oštećenje in vivo jeste nastanak ireverzibilnih oštećenja kože odnosno vidljive nekroze u epidermisu i dermisu nakon četvoročasovne primene ispitivane supstance. Korozivno oštećenje kože karakteriše se prema tipu izazvanog oštećenja kao: ulkus, krvarenje ili krvave kraste i na kraju perioda posmatranja od 14 dana prema promeni boje kože uslovljene perutanjem i pojavi površina sa alopecijom i ožiljnog tkiva. U cilju procene nejasnih oštećenja obavljaju se histopatološka ispitivanja.
Vijabilnost ćelija jeste parametar koji meri ukupnu aktivnost ćelijske populacije (npr. sposobnost ćelijske mitohondrijalne dehidrogenaze da smanji vitalnu boju MTT), koja, u zavisnosti od ciljnog pokazatelja ispitivanja koji se meri i korišćenog dizajna ispitivanja korelira sa ukupnim brojem i/ili vitalnosti ćelija.
Tabela 1: Referentne hemikalije
Naziv | EINECS broj | CAS broj |
|
1,2-diaminopropan | 201-155-9 | 78-90-0 | jako korozivno |
akrilna kiselina | 201-177-9 | 79-10-7 | jako korozivno |
2-terc-butilfenol | 201-807-2 | 88-18-6 | korozivno |
kalijum-hidroksid (10 %) | 215-181-3 | 1310-58-3 | korozivno |
sumporna kiselina (10 %) | 231-639-5 | 7664-93-9 | korozivno |
oktanska kiselina (kaprilna kiselina) | 204-677-5 | 124-07-02 | korozivno |
4-amino-1,2,4-triazol | 209-533-5 | 584-13-4 | nije korozivno |
eugenol | 202-589-1 | 97-53-0 | nije korozivno |
fenetil-bromid | 203-130-8 | 103-63-9 | nije korozivno |
tetrahloroetilen | 204-825-9 | 27-18-4 | nije korozivno |
izostearinska kiselina | 250-178-0 | 30399-84-9 | nije korozivno |
4-(metiltio)-benzaldehid | 222-365-7 | 3446-89-7 | nije korozivno |
Većina navedenih hemikalija preuzeto je sa spiska hemikalija izabranih za međunarodnu studiju validacije ECVAM4. Njihov izbor se bazira na sledećim kriterijumima:
a) jednak broj korozivnih i nekorozivnih supstanci;
b) komercijalno dostupne supstance koje pokrivaju većinu relevantnih hemijskih klasa;
v) obuhvatanje jako korozivnih kao i manje korozivnih supstanci kako bi se omogućilo razlikovanje na osnovu korozivnog potencijala;
g) izbor hemikalija kojima se može rukovati u laboratoriji bez postavljanja drugih ozbiljnih opasnosti osim korozivnosti.
Ispitivani materijal nanosi se površinski na trodimenzionalni model ljudske kože, koji se sastoji najmanje od rekonstruisanog epidermisa sa funkcionalnim stratum corneum-om. Korozivni materijali se identifikuju po njihovoj sposobnosti da izazovu smanjenje vijabilnosti ćelija (kao što je određeno npr. korišćenjem eseja smanjenja MTT15) ispod definisanih granica pri određenim periodima izlaganja. Princip ispitivanja modela humane kože zasniva se na hipotezi da korozivne hemikalije mogu da prodru kroz stratum corneum difuzijom ili erozijom i dovoljno su citotoksične da uzrokuju odumiranje potpornih slojeva ćelija.
1.4.1. Postupak
1.4.1.1. Modeli ljudske kože
Modeli ljudske kože mogu biti sastavljeni ili dobijeni komercijalno (npr. modeli EpiDermTM and EPISKINTM)16, 17, 18, 19 ili mogu biti razvijeni ili sastavljeni u laboratoriji koja vrši ispitivanje20, 21. Upotreba humane kože je predmet nacionalnih i međunarodnih etičkih razmatranja i uslova. Svaki novi model treba da bude validiran (najmanje u obimu koji je opisan u odeljku 1.4.1.1.2. ove metode). Modeli humane kože koji se koriste za ovo ispitivanje mora zadovoljavati sledeće:
1.4.1.1.1. Opšti uslovi za model
Humani keratinociti treba da se koriste za izradu epitelijuma. Ispod funkcionalnog stratum corneum-a treba da bude prisutno više slojeva vijabilnih epitelijalnih ćelija. Model kože može da ima sloj sa stromalnom komponentom. Stratum corneum treba da je višeslojan sa neophodnim lipidnim profilom da stvara funkcionalnu barijeru sa snagom da pruži otpor brzom prodiranju citotoksičnih markera. Svojstva sastava modela treba da spreče prolazak materijala oko stratum corneum-a do vijabilnog tkiva. Prolazak ispitivanih hemikalija oko stratum corneum-a dovodi do lošeg modelovanja ekspozicije kože. Model kože treba da bude bez kontaminacije bakterijama (uključujući mikoplazmu) ili gljivicama.
1.4.1.1.2. Uslovi za funkcionalni model
Veličina vijabilnosti obično se kvantifikuje upotrebom MTT ili druge vitalne boje koja se metabolički transformiše. U ovim slučajevima optička gustina (u daljem tekstu: OD) ekstrahovane (rastvorene) boje iz negativnog kontrolnog tkiva treba da je najmanje 20 puta veća od OD samog ekstrakcionog rastvarača (za opšti pregled videti literaturu22). Negativno kontrolno tkivo treba da bude stabilno u kulturi (obezbeđujući slična merenja vijabilnosti) u trajanju perioda ispitivanog izlaganja. Stratum corneum treba da bude dovoljno otporan da se odupre brzom prodiranju određenih hemikalija - citotoksičnih markera (tj. 1% Triton X-100). Ovo svojstvo može da bude procenjeno vremenom ekspozicije koje je potrebno da se smanji vijabilnost ćelija za 50% (ET50) (npr. za modele EpiDermTM and EPISKINTM ono je > 2 sata). Ovo tkivo treba da prikaže reproduktivnost tokom vremena i najbolje između laboratorija. Sem toga, ono treba da može da predvidi korozivni potencijal referentne hemikalije (videti Tabelu 1) kada se koristi u izabranom protokolu ispitivanja.
1.4.1.2. Primena ispitivanih i kontrolnih supstanci
Dva replikata (kopije) tkiva koriste se za svaki tretman (vreme izlaganja), uključujući kontrole. Za tečne materijale, dovoljno ispitivane supstance mora se naneti tako da ujednačeno prekrije površinu kože: treba koristiti najmanje 25 μL/cm2. Za čvrste materijale, dovoljno ispitivane supstance mora se naneti ujednačeno da pokrije kožu i treba je navlažiti sa dejonizovanom ili destilovanom vodom kako bi se omogućio dobar kontakt sa kožom. Kada je prikladno, čvrste supstance treba usitniti u prah pre primene. Metoda primene treba da odgovaraća za ispitivanu supstancu (videti literaturu5). Na kraju perioda izlaganja, ispitivani materijal se mora pažljivo oprati sa površine kože sa odgovarajućom puferom ili 0,9% NaCl.
Istovremene pozitivne i negativne kontrole treba koristiti za svaku studiju kako bi se obezbedila adekvatna performansa eksperimentalnog modela. Predložene supstance za pozitivnu kontrolu su glacijalna sirćetna kiselina ili 8N KOH. Predložene negativne kontrole su 0,9% NaCl ili voda.
1.4.1.3. Merenja vijabilnosti ćelija
Samo kvantitativne, validovane metode mogu se koristiti za merenje vijabilnosti ćelija. Mera vijabilnosti mora da bude kompatibilna sa upotrebom trodimenzionalne konstrukcije tkiva. Nespecifično vezivanje boje ne sme da interferira sa merenjem vijabiliteta. Boje koje se vezuju za protein i one koje ne podležu metaboličkoj transformaciji (npr. neutral red) zato nisu pogodne. Najčešće korišćeno ispitivanje je smanjenje MTT (3-(4,5-dimetiltiazol-2il)-2,5-difeniltetrazolijum bromid, Tiazol plavo: EINECS broj 206-069-5, CAS broj 298-93-1)), za koji je pokazano da daje tačne i reproduktivne rezultate5, ali mogu se koristiti i druga ispitivanja. Uzorak kože se stavlja u rastvor MTT odgovarajuće koncentracije (npr. 0,3 - 1 mg/mL) na odgovarajućoj inkubacionoj temperaturi tokom tri sata. Istaloženi plavi proizvod formazan se zatim ekstrahuje upotrebom rastvarača (izopropanola) i meri se koncentracija formazana se meri određivanjem OD na talasnoj dužini između 540 nm i 595 nm.
Hemijsko dejstvo ispitivanog materijala na vitalnu boju može da liči na metabolizam ćelije vodeći do pogrešne procene vijabilnosti. Ovo se dešava kada takav ispitivani materijal nije potpuno uklonjen sa kože ispiranjem9. Ako ispitivani materijal direktno deluje na vitalnu boju, dodatne kontrole treba koristiti kako bi se detektovala i korigovala interferencija ispitivane supstance sa merenjem vijabiliteta9, 23.
Za svako tkivo navode se OD vrednosti i izračunati procenti vijabilnosti ćelija za svaki ispitivani materijal, pozitivne i negativne kontrole, uključujući podatke od replikata ponovljenih eksperimenata kako odgovara, srednje i pojedinačne vrednosti.
Dobijene OD vrednosti za svaki ispitivani uzorak mogu se koristiti za izračunavanje procenta vijabiliteta u odnosu na negativnu kontrolu, koja je 100%. Granična vrednost procenta vijabliteta koja pravi razliku između korozivnih i nekorozivnih materijala (ili razliku između različitih klasa koroziva) ili statistička procedura koja se koristi da se ocene rezultati i identifikuju korozivni materijali, moraju se jasno definisati i dokumentovati i prikazati da su odgovarajući. Ove granične vrednosti utvrđuju se za vreme optimizacije ispitivanja, ispituju se tokom prevalidacione faze i potvrđuju u validacionoj studiji. Kao primer predviđanje korozivnosti koja je u vezi sa modelom EpiDermTM je9:
Ispitivana supstanca se smatra korozivnom za kožu:
a) ako je vijabilnost posle tri minuta izlaganja manja od 50% ili
b) ako je vijabilnost posle tri minuta izlaganja veća ili jednaka 50% i vijabilnost posle jednog sata izloženosti manja od 15%.
Ispitivana supstanca se ne smatra korozivnom za kožu:
a) ako je vijabilnost posle tri minuta izloženosti veća ili jednaka 50% i vijabilnost posle 1 sata izlaganja veća ili jednaka 15%.
Izveštaj o ispitivanju, ukoliko je moguće, sadrži podatke o:
1) Ispitivanim i kontrolnim supstancama:
- hemijsko ime kao što je IUPAC ili CAS ime i CAS broj, ako je poznato;
- čistoća i sastav supstance ili smeše (u težinskim procentima);
- fizičke i hemijske osobine kao što su fizičko stanje, pH, stabilnost, rastvorljivost u vodi, ono što je relevantno za izvođenje studije;
- tretman ispitivane/kontrolne supstance pre ispitivanja, ako je primenljivo (npr. zagrevanje, usitnjavanje);
- stabilnost, ako je poznata.
2) Opravdanju modela kože i korišćenog postupka.
3) Uslovima ispitivanja:
- korišćen sistem ćelija;
- podatke o kalibraciji za uređaj za merenje koji se koristi za merenje vijabilnosti ćelija (npr. spektrofotometar);
- svi podaci na koje se oslanjalo za specifični model kože uključujući njegovu validaciju;
- detalji postupka ispitivanja;
- korišćene doze;
- opis modifikacije postupka ispitivanja;
- reference prema istorijskim podacima za model;
- opis korišćenih kriterijuma za ocenjivanje.
4) Rezultatima:
- podaci pojedinačnih ispitivanih uzoraka u tabeli;
- opis drugih primećenih efekata.
5) Obrazloženju rezultata.
6) Zaključcima.
1. OECD (2001) Harmonised Integrated Classification System for Human Health and Environmental Hazards of Chemical Substances and Mixtures. OECD Series on Testing and Assessment Number 33. ENV/JM/MONO(2001)6, Paris. http://www.olis.oecd.org/olis/2001doc.nsf/LinkTo/env-jm-mono(2001)6.
2. Testing Method B.4. Acute Toxicity: Dermal Irritation/Corrosion.
3. Botham, P.A., Chamberlain, M., Barratt, M.D., Curren, R.D., Esdaile, D.J., Gardner, J.R., Gordon, V.C., Hildebrand, B., Lewis, R.W., Liebsch, M., Logemann, P., Osborne, R., Ponec, M., Regnier, J.F., Steiling, W., Walker, A.P., and Balls, M., (1995) A prevalidation study on in vitro skin corrosivity testing. The report recommendations of ECVAM Workshop 6 ATLA 23, p. 219-255.
4. Barratt, M.D., Brantom, P.G., Fentem, J.H., Gerner, I., Walker, A.P., and Worth, A.P. (1998). The ECVAM international validation study on in vitro tests for skin corrosivity. 1. Selection and distribution of the test chemicals. Toxicology In vitro 12, p. 471-482.
5. Fentem, J.H., Archer, G.E.B., Balls, M., Botham, P.A., Curren, R.D., Earl, L.K., Esdaile, D.J., Holzhutter, H.G. and Liebsch, M., (1998) The ECVAM international validation study on in vitro tests for skin corrosivity. 2. Results and evaluation by the Management Team. Toxicology In vitro 12, p. 483-524.
6. OECD, (1996) Final Report of the OECD Workshop on Harmonisation of Validation and Acceptance Criteria for Alternative Toxicological Test Methods, p. 62.
7. Balls, M., Blaauboer, B.J., Fentem, J.H., Bruner, L., Combes, R.D., Ekwall, B., Fielder, R.J., Guillouzo, A., Lewis, R.W., Lovell, D.P., Reinhardt, C.A., Repetto, G., Sladowski, D., Spielmann, H., and Zucco, F., (1995) Practical aspects of the validation of toxicity test procedures. Test report and recommendations of ECVAM workshops. ATLA 23, p. 129-147.
8. ICCVAM (Interagency Coordinating Committee on the Validation of Alternative Methods)., (1997) Validation and Regulatory Acceptance of Toxicological Test Methods. NIH Publication No 97-3981. National Institute of Environmental Health Sciences, Research Triangle Park, NC, USA. http://iccvam.niehs.nih.gov/docs/guidelines/validate.pdf.
9. Liebsch, M., Traue, D., Barrabas, C., Spielmann, H., Uphill, P., Wilkins, S., McPherson, J.P., Wiemann, C., Kaufmann, T., Remmele, M. and Holzhutter, H.G., (2000) The ECVAM prevalidation study on the use of EpiDerm for skin Corrosivity testing. ATLA 28, p. 371-401.
10. ECVAM, (1998) ECVAM News & Views. ATLA 26, p. 275-280.
11. ECVAM, (2000) ECVAM News & Views. ATLA 28, p. 365-67.
12. ICCVAM (Interagency Coordinating Committee on the Validation of Alternative Methods)., (2002) ICCVAM evaluation of EpiDermTM, EPISKINTM (EPI-200), and the Rat Skin Transcutaneous Electrical Resistance (TER) assay: In vitro test methods for assessing dermal corrosivity potential of chemicals. NIH Publication No 02-4502. National Toxicology Program Interagency Center for the Evaluation of Alternative Toxicological Methods, National Institute of Environmental Health Sciences, Research Triangle Park, NC, USA. http://iccvam.niehs.nih.gov/methods/epiddocs/epis_brd.pdf.
13. OECD, (2002) Extended Expert Consultation Meeting on The In vitro Skin Corrosion Test Guideline Proposal, Berlin, 1st-2nd November 2001, Secretariat's Final Summary Report, 27th March 2002, OECD ENV/EHS, available upon request from the Secretariat
14. Worth, A.P., Fentem, J.H., Balls, M., Botham, P.A., Curren, R.D., Earl, L.K., Esdaile, D.J., Liebsch, M., (1998) An Evaluation of the Proposed OECD Testing Strategy for Skin Corrosion. ATLA 26, p. 709-720.
15. Mosmann, T., (1983) Rapid colorimetric assay for cellular growth and survival: application to proliferation and cytotoxicity asssays. J.Immunol. Meth. 65, p. 55-63.
16. Cannon, C.L., Neal, P.J., Southee, J.A., Kubilus, J., and Klausner, M., 1994. New epidermal model for dermal irritancy testing. Toxicology In vitro 8, p. 889-891.
17. Ponec, M., Boelsma, E., Weerheim, A., Mulder, A., Boutwstra, J., and Mommaas, M., 2000. Lipid and ultrastructural characterisation of reconstructed skin models. International Journal of Pharmaceutics. 203, p. 211-225.
18. Tinois, E., Gaetani, Q., Gayraud, B., Dupont, D., Rougier, A., Pouradier, D.X., (1994) The Episkin model: Successful reconstruction of human epidermis in vitro. In In vitro Skin Toxicology. Edited by A Rougier, AM Goldberg and HI Maibach, p. 133-140
19. Tinois E, Tiollier J, Gaucherand M, Dumas H, Tardy M, Thivolet J (1991). In vitro and post-transplantation differentiation of human keratinocytes grown on the human type IV collagen film of a bilayered dermal substitute. Experimental Cell Research 193, p. 310-319
20. Parentau, N.L., Bilbo, P., Molte, C.J., Mason, V.S., and Rosenberg, H., (1992) The organotypic culture of human skin keratinocytes and fibroblasts to achieve form and function. Cytotechnology 9, p. 163-171.
21. Wilkins, L.M., Watson, S.R., Prosky, S.J., Meunier, S.F., Parentau, N.L., (1994) Development of a bilayered living skin construct for clinical applications. Biotechnology and Bioengineering 43/8, p. 747-756.
22. Marshall, N.J., Goodwin, C.J., Holt, S.J., (1995) A critical assessment of the use of microculture tetrazolium assays to measure cell growth and function. Growth Regulation 5, p. 69-84.
23. Fentem, J.H., Briggs, D., Chesne', C., Elliot, G.R., Harbell, J.W., Heylings, J.R., Portes, P., Rouget, R., and van de Sandt, J.J.M., and Botham, P.A., (2001) A prevalidation study on in vitro tests for acute skin irritation: results and evaluation by the Management Team. Toxicology In vitro 15, p. 57-93.
B.41. IN VITRO 3T3 NRU ISPITIVANJE FOTOTOKSIČNOSTI
Ova metoda potpuno odgovara metodi OECD: TG 432 (2004).
Fototoksičnost se definiše kao toksična reakcija na supstancu primenjenu na organizam koja je ili izazvana ili pojačana (jasna pri nižim doznim nivoima) posle naknadnog izlaganja svetlosti ili koja je uzrokovana zračenjem kože posle sistemske primene supstance.
In vitro 3T3 NRU ispitivanje fototoksičnosti se koristi za identifikovanje fototoksičnog potencijala ispitivane supstance koji je izazvan pobuđenom hemikalijom posle ekspozicije svetlu. Ispitivanje ocenjuje foto-citotoksičnost relativnim smanjenjem u vijabilnosti ćelija koje su izložene hemikaliji u prisustvu svetlosti u odnosu na odsustvo svetlosti. Supstance koje su identifikovane ovim ispitivanjem su verovatno fototoksične in vivo po sistemskoj primeni i distribuciji u kožu ili posle površinske primene (lokalne primene na kožu).
Mnoge vrste hemikalija su prijavljene da izazivaju fototoksične efekte1, 2, 3, 4. Njihova zajednička osobina je njihova sposobnost da apsorbuju svetlosnu energiju u opsegu sunčeve svetlosti. Prema prvom zakonu fotohemije (zakon Grotthaus-Draper), fotoreakcija zahteva dovoljnu apsorpciju kvantuma svetlosti. Zbog toga, pre razmatranja biološkog ispitivanja, treba odrediti UV/vis apsorpcioni spektar ispitivane supstanceLI. Predloženo je da ako je molarni ekstinkcioni/apsorpcioni koeficijent manji od 10 litre x mol-1 x cm-1 onda supstanca verovatno nije fotoreaktivna. Takvu hemikaliju nije neophodno ispitivati in vitro 3T3 NRU ispitivanjem fototoksičnosti ili bilo kojim drugim ispitivanjem za štetne fotohemijske efekte1, 5. Videti Deo drugi ove metode.
Pouzdanost i relevantnost in vitro 3T3 NRU ispitivanja fototoksičnosti je nedavno evaluirana6, 7, 8, 9. In vitro 3T3 NRU test fototoksičnosti se pokazao predvidivim za akutne fototoksične efekte kod životinja i ljudi in vivo. Test nije dizajniran da predvidi druge štetne efekte koji bi se mogli pojaviti iz kombinovanog dejstva hemikalije i svetlosti, npr. ne odnosi se na fotogenotoksičnost, fotoalergiju ili fotokarcinogenost, niti dozvoljava procenu potencijala fototoksičnosti. Test nije dizajniran da se odnosi na indirektne mehanizme fototoksičnosti, efekte metabolita ispitivane supstance ili efekte smeša.
Kako je upotreba metabolišućih sistema opšti zahtev za sve in vitro testove za predviđanje genotoksičnosti i karcinogenog potencijala, do sada, u slučaju fototoksikologije, postoje samo retki primeri kada je potrebna metabolička transformacija za hemikaliju da bi se ponašala kao fotootrov in vivo ili in vitro. Zbog toga se ne smatra neophodnim niti naučno opravdanim za ovaj test da se izvodi sa sistemom metaboličke aktivacije.
______________
LI Prema OECD TG 101
Zračenje jeste intenzitet upadnog ultravioletnog (u daljem tekstu: UV) ili vidljivog svetla na površini, meren u W/m2 ili mW/cm2.
Doza svetlosti jeste količina (koja je jednaka proizvodu intenziteta i vremena) UV ili vidljivog upadnog zračenja na površini, izražen u džulima (= W x s) po jedinci površine, npr. J/m2 ili J/cm2.
Talasne dužine UV svetlosti jesu oznake koje je preporučila CIE (Commission Internationale de L'Eclairage), i to su: UVA (315 nm do 400 nm), UVB (280 nm do 315 nm) i UVC (100 nm do 280 nm). Druge oznake su takođe u upotrebi: podela između UVB i UVA se često stavlja na 320 nm, a UVA može biti podeljena u UV-A1 i UV-A2 s podelom napravljenom na oko 340 nm.
Viabilitet ćelija jeste parametar koji meri ukupnu aktivnost ćelijske populacije (npr. upijanje vitalne boje Neutral Red u ćelijske lizozome), koja u zavisnosti od ciljnog pokazatelja ispitivanja koji se meri i korišćenog dizajna ispitivanja korelira sa ukupnim brojem i/ili vitalnosti ćelija.
Relativni viabilitet ćelija jeste ćelijski viabilitet izražen u odnosu na negativne (rastvarač) kontrole koje su prošle celi eksperimentalni postupak (+Irr ili -Irr), ali nisu tretirane sa ispitivanom hemikalijom.
Fotoiritacioni faktor (u daljem tekstu: PIF) jeste faktor izveden upoređivanjem dve podjednako efikasne citotoksične koncentracije (IC50) ispitivane hemikalije dobijene u odsustvu (-Irr) i u prisustvu (+Irr) necitotoksičnog zračenja sa UVA/vis svetlošću.
IC50 jeste koncentracija ispitivane hemikalije koja smanjuje viabilnost ćelija za 50%.
Srednji fotoefekat (u daljem tekstu: MPE) jeste mera izvedena iz matematičke analize krivih koncentracija odgovor dobijenih u odsustvu (-Irr) i u prisustvu (+Irr) zračenja sa UVA/vis svetlošću koje nije citotoksično.
Fototoksičnost jeste akutna toksična reakcija koja je izazvana posle prvog izlaganja kože određenim hemikalijama i naknadnom izlaganju svetlu ili koja je slično izazvana zračenjem kože posle sistemske primene hemikalije.
In vitro 3T3 NRU test fototoksičnosti zasnovan je na poređenju citotoksičnosti hemikalije, kada se ispituje u prisustvu i odsustvu izlaganja necitotoksičnoj dozi simulirane sunčeve svetlosti. Citotoksičnost je u ovom testu izražena kao smanjenje preuzimanja vitalne boje, Neutral Red. Zavisno od koncentracije, kada se meri 24 sata posle tretmana ispitivanom hemikalijom i zračenja10. NR je slaba katjonska boja koja lako prodire kroz membrane ćelija ne-difuzijom, akumulirajući se intracelularno u lizosomima. Promene u površini osetljive lizosomalne membrane vodi do lizosomalne nepostojanosti i drugih promena koje postepeno postaju ireverzibilne. Takve promene koje su nastale dejstvom ksenobiotika imaju za posledicu smanjenje preuzimanja i vezivanja NR. Na ovaj način se može napraviti razlika između viabilnih, oštećenih ili mrtvih ćelija, što je osnova ovog ispitivanja.
Balb/c 3T3 ćelije se održavaju u kulturi 24 sata da bi se formirali monoslojevi. Dve posude sa 96 bazenčića po ispitivanoj hemikaliji se pre-inkubiraju sa osam različitih koncentracija ispitivane supstance u trajanju od 1 sata. Potom se jedna od dve posude izlaže najvišoj dozi ne-citotoksičnog zračenja dok se druga posuda drži u mraku. Zatim se u obe posude medijum tretmana zameni medijumom za kultivisanje i posle sledećih 24 sata inkubacije određuje se viabilitet ćelija preuzimanjem Neutral Red boje. Viabilitet ćelija se izražava kao procenat od netretiranih kontrola rastvarača i računa se za svaku ispitivanu koncentraciju. Kako bi se predvideo potencijal fototoksičnosti, porede se koncentracione reakcije dobijene u prisustvu i u odsustvu zračenja, obično pri nivou IC50, tj. pri koncentraciji koja smanjuje viabilitet ćelija do 50% u poređenju sa netretiranim kontrolama.
1.4.1. Pripreme
1.4.1.1. Ćelije
Permanentna ćelijska linija mišjih firoblasta, Balb/c 3T3, klon 31, bilo iz Kolekcije kultura američkog tipa (American Type Culture Collection, ATCC), Manassas, VA, USA ili iz Evropske Kolekcije ćelijskih kultura (The European Collection of Cell Cultures, ECACC, Salisbury, Wiltshire, UK) korišćena je u studiji validacije i zato se preporučuje da se nabave iz dobro kvalifikovanih depoa ćelija. Uz isti postupak ispitivanja mogu se koristiti druge ćelije ili ćelijske linije ako su uslovi kultivisanja prilagođeni specifičnim potrebama ćelija, ali se mora pokazati ekvivalentnost.
Ćelije treba redovno proveravati na odsustvo kontaminacije mikoplazmom i koristiti jedino ako nijedna mikoplazma nije pronađena11.
Važno je da se redovno proverava osetljivost ćelija na UV prema postupcima kontrole kvaliteta koje su opisane u ovoj metodi. Zbog toga što se UVA osetljivost ćelija može povećati s brojem prolaza, treba koristiti Balb/c 3T3 ćelije najnižeg broja prolaza koji se može dobiti, najbolje manje od 100 (videti odeljak 1.4.2.2.2. ove metode i Deo treći ove metode).
1.4.1.2. Medijum i uslovi kultivisanja
Odgovarajući medijum za kultivisanje i uslovi kultivacije treba da se koriste za rutinske prolaze ćelija i tokom postupka ispitivanja, npr. za ćelije Balb/c 3T3 oni su DMEM (Dulbecco's Modified Eagle's Medium) kome je dodato 10% seruma novorođenog govečeta, 4 mM glutamina, penicilin (100 IU) i streptomicin (100 μg/mL), i vlažna inkubacija na 37 °C, 5% - 7,5% CO2 u zavisnosti od pufera (videti odeljak 1.4.1.4. drugi pasus ove metode). Naročito je važno da uslovi kultivisanja ćelija obezbede da vreme ciklusa ćelija bude u okviru normalnog istorijskog opsega ćelija ili ćelijskih linija koje se koriste.
1.4.1.3. Pripremanje kultura
Ćelije iz zamrznutih "stock" kultura zaseju se u medijum za kultivisanje pri odgovarajućoj gustini i subkultivišu najmanje jednom pre upotrebe u in vitro 3T3 NRU testu fototoksičnosti.
Ćelije koje se koriste za test fototoksičnosti poseju se u medijum za kultivisanje pri odgovarajućoj gustini tako da kulture ne dostignu nagomilavanje do kraja testa, tj. kada je viabilitet ćelija određen 48 sati posle zasejavanja ćelija. Za ćelije Balb/c 3T3 koje rastu u posudama od 96 bazenčića, preporučena gustina zasejavanja ćelija je 1 x 104 ćelija po bazenčiću.
Za svaku ispitivanu supstancu ćelije se zaseju indentično u dve odvojene posude sa 96 bazenčića, koji zatim istovremeno prolaze postupak ispitivanja pod identičnim uslovima kultivacije osim za vremenski period kada se jedna posuda izlaže zračenju (+Irr) a druga se drži u mraku (-Irr).
1.4.1.4. Priprema ispitivane supstance
Ispitivane supstance se moraju pripremiti sveže, odmah pre upotrebe osim ukoliko podaci pokazuju njihovu stabilnost pri čuvanju. Preporučuje se da se celokupna priprema hemikalija i inicijalni tretman ćelija izvede pod svetlosnim uslovima koji bi izbegli fotoaktivaciju ili degradaciju ispitivane supstance ispitivane supstance pre zračenja.
Ispitivane hemikalije treba rastvoriti u puferovanim rastvorima soli, npr. Earle's Balanced Salt Solution (EBSS) ili drugom fiziološki izbalansiranom puferskom rastvoru koji ne sme da sadrži proteinske sastojke, sastojke koji apsorbuju svetlost (npr. pH-idikatorke boje ili vitamini) kako bi se izbegla interferencija tokom zračenja. Kako se tokom zračenja ćelije drže oko 50 minuta van CO2 inkubatora, mora se voditi računa da se izbegne alkalizacija. Ako se koriste slabi puferi kao EBSS, ovo se može postići inkubiranjem ćelija pri 7,5% CO2. Ako se ćelije inkubiraju pri samo 5% CO2, treba izabrati jači pufer.
Ispitivane hemikalije ograničene rastvorljivosti u vodi treba rastvoriti u odgovarajućem rastvaraču. Ako se koristi rastvarač, on mora da bude prisutan u konstantnoj zapremini u svim kulturama, tj. u negativnoj kontroli (kontroli rastvarača) kao i u svim koncentracijama ispitivane supstance i da ne bude toksičan pri tim koncentracijama. Koncentracije ispitivane supstance treba izabrati tako da se izbegne taloženje ili mutnoća rastvora.
Preporučeni rastvarači su dimetilsulfoksid (u daljem tekstu: DMSO) i etanol (u daljem tekstu: ETOH). Mogu odgovarati i drugi rastvori niske citotoksičnosti. Pre upotrebe sve rastvarače treba proceniti prema specifičnim osobinama, npr. reakcija sa ispitivanom supstancom, gušenje fototoksičnog efekta, osobine dejonizacije radikala i/ili hemijska stabilnost u rastvaraču.
Mešanje na vorteksu i/ili zagrevanje do odgovarajućih temperatura može da se koristi kako bi se pomoglo rastvaranje ukoliko ovo ne bi imalo uticaja na stabilnost ispitivane supstance.
1.4.1.5. Uslovi zračenja
1.4.1.5.1. Izvor svetlosti
Izbor odgovarajućeg izvora svetlosti i filtera je kritičan faktor u ispitivanju fototoksičnosti. Svetlost UVA i vidljive oblasti obično se dovodi u vezu sa fototoksičnim odgovorima in vivo3, 12, dok je generalno UVB od manjeg značaja ali je visoko citotoksična. Citotoksičnost se povećava 1.000 puta kako talasna dužina ide od 313 nm do 280 nm13. Kriterijumi za izbor odgovarajućeg izvora svetlosti moraju da uključe zahtev da svetlosni izvor emituje talasne dužine koje apsorbuje ispitivana supstanca (apsorpsioni spektar) i da doza svetlosti (koja se može postići u razumnom vremenu izlaganja) treba da bude dovoljna za detekciju poznatih fotocitotoksičnih hemikalija. Talasne dužine i doze koje se koriste ne treba da prekomerno štete ispitivanom sistemu, npr. oslobađanje toplote (infracrvena oblast).
Simulacija sunčeve svetlosti sa solarnim simulatorima smatra se optimalnim veštačkim izvorom svetlosti. Raspodela snage zračenja filtriranog solarnog simulatora treba da bude približna dnevnoj svetlosti spoljašnjeg prostora koja je data u literaturi14. Ksenonski luk i (dopirani) živa-metal halogeni luk se koriste kao solarni simulatori15. Druga ima prednost emitovanja manje toplote i što je jeftinija slaganje sa sunčevom svetlošću je manje idealna u poređenju sa ksenonskim lukovima. Zbog toga što svi solarni simulatori emituju značajne količine UVB one treba da se odgovarajuće filtriraju da bi se prigušile UVB talasne dužine visoke citotoksičnosti. Zbog toga što plastični materijali za kultivisanje ćelija sadrže UV stabilizatore spektar bi trebalo meriti kroz isti tip poklopca posude sa 96 bazenčića kao onaj koji će se koristiti u testu. Nezavisno od mera preduzetih da bi se prigušili delovi spektra filtriranjem ili neizbežnim filterskim efektima opreme, spektar zabeležen ispod ovih filtera ne treba da odstupa od standardizovane spoljašnje dnevne svetlosti14. Primer distribucije spektralnog zračenja filtriranog solarnog simulatora korišćenog u studiji validacije in vitro 3T3 NRU testa fototoksičnosti je dat u literaturi8, 16. Videti Deo treći Slika 1. ove metode.
1.4.1.5.2. Doziranje
Intenzitet svetlosti (zračenja) treba redovno proveravati pre svakog testa fototoskičnosti koristeći odgovarajući UV-metar širokog opsega. Intenzitet treba meriti preko istog tipa poklopca posude sa 96 bazenčića kao što će se koristiti u testu. UV-metar mora da bude kalibrisan prema izvoru. Treba proveriti karakteristike UV-metra i u ovu svrhu preporučuje se upotreba drugog, referentnog UV-metra iste vrste i ista kalibracija. Idealno, pri većim intervalima treba koristiti spektroradiometar za merenje spektralnog zračenja filtriranog izvora svetlosti i proveriti kalibraciju UV-metra širokog opsega.
Doza od 5 J/cm2 (kao što je merena u UVA opsegu) je određena da ne bude citotoksična za ćelije Balb/c 3T3 i dovoljno jaka da pobudi hemikalije da izazovu fototoksičnu reakciju6, 7 npr. da postigne 5 J/cm2 tokom vremenskog perioda od 50 minuta zračenje je bilo podešeno na 1,7 mW/cm2. Videti Deo treći Slika 2. ove metode. Ukoliko se koristi druga ćelijska linija ili drugi svetlosni izvor, doza zračenja možda treba da se kalibriše tako da način doziranja može da bude izabran da nije štetan za ćelije ali da je dovoljan da pobudi standardne fotootrove. Vreme izlaganja svetlosti se računa na sledeći način:
t(min) = | doza zračenja (J / cm2) x 100 | |
zračenje (mW / cm2) x 60 |
(1 J = 1 Wsec)
1.4.2. Eksperimentalni uslovi
1.4.2.1. Koncentracije ispitivane supstance
Opsezi koncentracija ispitivane hemikalije u prisustvu (+Irr) ili u odsustvu (-Irr) svetlosti treba da budu adekvatno određeni u eksperimentima koji su osmišljeni za nalaženje opsega. Bilo bi korisno prvo proceniti rastvorljivost i na 60 minuta (ili koje vreme će se koristiti) jer rastvorljivost može da se promeni tokom vremena ili za vreme trajanja ekspozicije. Da bi se izbegla toksičnost koja je uzrokovana neodgovarajućim uslovima kultivisanja ili visokom kiselošću ili alkalnosti hemikalije, pH ćelijskih kultura sa dodatom ispitivanom supstancom treba da bude u opsegu 6,5 do 7,8.
Najviša koncentracija ispitivane supstance treba da bude u okviru fizioloških uslova ispitivanja, npr. treba da se izbegne osmotski ili pH stres. U zavisnosti od ispitivane hemikalije, možda je neophodno razmotriti druge fizikohemijske osobine kao ograničavajuće faktore za najvišu ispitivanu koncentraciju. Za relativno nerastvorne supstance koje nisu toksične pri koncentracijama do tačke zasićenja treba ispitati najvišu koncentraciju koja se može postići.
Treba da se izbegne taloženje ispitivane hemikalije pri bilo kojoj ispitivanoj koncentraciji. Maksimalna koncentracija ispitivane supstance ne treba da prelazi 1.000 μg/mL; osmolalnost ne treba da prelazi 10 mmolar. Treba da se koristi serija geometrijskog razblaženja osam koncentracija ispitivane supstance sa konstantnim faktorom razblaženja (videti odeljak 2.1. drugi pasus ove metode).
Ukoliko postoje podaci (iz eksperimenta nalaženja opsega) da ispitivana supstanca nije citotoksična do granice koncentracije u eksperimentu u mraku (-Irr), ali je visoko toksična kada je ozračena (+Irr), opseg koncentracija koji se bira za (+Irr) eksperiment može da se razlikuje od onog izabranog za (−Irr) eksperiment da bi ispunio zahtev adekvatnosti kvaliteta podataka.
1.4.2.2. Kontrole
1.4.2.2.1. Osetljivost ćelija na zračenje, uspostavljanje istorijskih podataka
Ćelije treba proveravati redovno (približno svakog petog prolaza) na osetljivost prema izvoru svetlosti procenom njihove viabilnosti po ekspoziciji rastućim dozama zračenja. U ovoj proceni treba da se koristi nekoliko doza zračenja, uključujući nivoe značajno veće od onih koji se koriste u 3T3 NRU testu fototoksičnosti. Ove doze se najlakše kvantifikuju merenjima UV delova izvora svetlosti. Ćelije se zasade pri gustini koja se koristi u in vitro 3T3 NRU testu fototoksičnosti i zrače se sledećeg dana. Onda se određuje viabilitet ćelija dan kasnije korišćenjem preuzimanja Neutral Red boje. Treba pokazati da je nastala najveća necitotoksična doza (npr. u studiji validacije: 5 J/cm2 (UVA)) bila dovoljna da ispravno klasifikuje referentnu hemikaliju (Tabela 1).
1.4.2.2.2. Osetljivost na zračenje, provera tekućeg testa
Ispitivanje zadovoljava kriterijume kvaliteta ako ozračene negativne kontrole/kontrole rastvarača pokažu viabilitet veći od 80% u poređenju sa neozračenom negativnom kontrolom/kontrolom rastvarača.
1.4.2.2.3. Sposobnost rasta kontrola rastvarača
Apsolutna optička gustina (OD540 NRU) boje Neutral Red ekstrahovane iz kontrole rastvarača ukazuje na to da li je 1x104 ćelija koje su zasejane po bazenčiću pokazalo rast sa normalnim vremenom udvostručavanja tokom dva dana testa. Ispitivanje zadovoljava kriterijume prihvatanja ako je srednja vrednost OD540 NRU netretiranih kontrola ≥0,4 (tj. približno 20 puta veća od podloge apsorbancije rastvarača).
1.4.2.2.4. Pozitivna kontrola
Poznata fototoksična hemikalija treba da se testira istovremeno sa svakim in vitro 3T3 NRU testom fototoksičnosti. Preporučuje se hlorpromazin (u daljem tekstu: CPZ). Za CPZ koji se testira sa standardnim postupkom u in vitro 3T3 NRU testu fototoksičnosti, određeni su kriterijumi za prihvatanje testa: ozračen CPZ (+Irr): IC50=0,1 do 2,0μg/ml, neozračen CPZ (-Irr): IC50 = 7,0 do 90,0 μg/mL. Foto iritacioni faktor (Photo Irritation Factor, PIF) treba da bude >6. Treba pratiti istorijske karakteristike pozitivne kontrole.
Mogu da se koriste druge fototoksične hemikalije, koje odgovaraju za hemijsku klasu ili karakteristike rastvorljivosti hemikalije koja se procenjuje, kao istovremena pozitivna kontrola umesto hlorpromazina.
1.4.3. Postupak ispitivanja6, 7, 8, 16, 17
1.4.3.1. Prvi dan
Razdeliti 100 μL medijuma za kultivisanje u periferne bazenčiće posude sa 96 bazenčića za mikrotitraciju tkivnih kultura (= slepe probe). U preostale bazenčiće, razdeliti 100 μL suspenzije ćelija od 1x105 ćelija/mL u medijumu za kultivisanje (= 1×104 ćelija/bazenčić). Dve posude treba pripremiti za svaku seriju pojedinačnih koncentracija ispitivane supstance i za kontrolu rastvarača i konpozitivnu kontrolu.
Inkubirati ćelije 24 sata (videti odeljak 1.4.1.2. ove metode) dok ne formiraju polukonfluentan monosloj. Ovaj period inkubacije dozvoljava oporavak ćelija, adherenciju i eksponencijalni rast.
1.4.3.2. Drugi dan
Posle inkubacije dekantovati medijum kulture od ćelija i isprati pažljivo sa 150 μL puferovanog rastvora koji je korišćen za inkubaciju. Dodati 100 μL pufera koji sadrži odgovarajuću koncentraciju ispitivane supstance ili rastvarača (kontrola rastvarača). Primeniti osam različitih koncentracija ispitivane hemikalije. Inkubitari ćelije sa ispitivanom supstancom u mraku 60 minuta (videti odeljak 1.4.1.2. i odeljak 1.4.1.4. drugi pasus ove metode).
Od dve posude pripremljene za svaku seriju koncentracija ispitivane supstance i kontrola, jedna se izabere, obično nasumično, za određivanje citotoksičnosti (-Irr) (t.j. kontrolna posuda) i jedna (tretirana posuda) za određivanje fototoksičnosti (+Irr).
Za izvođenje +Irr ekspozicije, ozračiti ćelije na sobnoj temperaturi 50 minuta preko poklopca posudice sa 96 bazenčića sa najvišom dozom zračenja koja nije citotoksična (videti Deo treći ove metode). Držati neozračene posude (-Irr) na sobnoj temperaturi u mračnoj kutiji 50 minuta (= vreme izlaganja svetlosti).
Dekantovati ispitivani rastvor i pažljivo isprati dva puta sa 150 μL puferskog rastvora koji je korišćen za inkubaciju, ali koji ne sadrži ispitivani materijal. Zameniti pufer sa medijumom za kultivaciju i inkubirati (videti odeljak 1.4.1.2. ove metode) preko noći (18 do 22 sata).
1.4.3.3. Treći dan
1.4.3.3.1. Mokroskopska procena
Ćelije treba pregledati na rast, morfologiju i integritet monosloja koristeći fazno kontrasni mikroskop. Beleže se promene u morfologiji ćelija i efekti na ćelijski rast.
1.4.3.3.2. Test preuzimanja boje Neutral Red
Isprati ćelije sa 150 μL prethodno zagrejanog pufera. Ukloniti rastvor za ispiranje nežnim lupkanjem. Dodati 100 μL rastvora Neutral Red (u daljem tekstu: NR) (3-amino-7-dimetilamino-2-metilfenazin hidrohlorid, EINECS broj 209-035-8; CAS broj 553-24-2; C.I. 50040) koncentracije 50 μg/mL u medijum bez seruma16 i inkubirati kao što je opisano u odeljku 1.4.1.2. ove metode tokom tri sata. Posle inkubacije, ukloniti NR medijum i isprati ćelije sa 150 μL pufera. Dekantovati i ukloniti višak pufera upijanjem ili centrifugiranjem.
Dodati tačno 150 μL rastvora za odstranjivanje NR (sveže pripremljen 49 delova vode + 50 delova etanola + 1 deo sirćetne kiseline).
Mešati posudu za mikrotitraciju nežno na mešalici 10 minuta dok se NR ne ekstrahuje iz ćelija i formira homogeni rastvor.
Izmeriti optičku gustinu ekstrakta NR na 540 nm na spektrofotometru prema slepoj probi. Sačuvati podatke u dokumentu odgovarajućeg elektronskog formata za dalju analizu.
2.1. KVALITET I KVANTITET PODATAKA
Podaci ispitivanja treba da omoguće značajnu analizu koncentracije-odgovora dobijene u prisustvu i u odsustvu zračenja i, ako je moguće, koncentraciju ispitivane hemikalije pri kojoj je viabilnost ćelija smanjena na 50% (IC50). Ukoliko se pronađe citotoksičnost i opseg koncentracija i odsečak pojedinačnih koncentracija treba da budu postavljeni tako da omoguće podešavanje krive prema eksperimentalnim podacima.
Za jasno pozitivne i za jasno negativne rezultate (vedeti odeljak 2.3. prvi pasus ove metode), može da bude dovoljan primarni eksperiment koji je podržan jednim ili sa više preliminarnih ispitivanja nalaženja opsega.
Dvosmisleni, granični ili nejasni rezultati treba da budu razjašnjeni daljim ispitivanjima (videti odeljak 2.4. drugi pasus ove metode). U takvim slučajevima treba razmotriti izmenu eksperimentalnih uslova. Eksperimentalni uslovi koji se mogu izmeniti uključuju opseg koncentracija ili razmak između koncentracija, vreme pre-inkubacije i vreme izlaganja zračenju. Kraće vreme izlaganja može da bude pogodno za hemikalije nestabilne u vodi.
Da bi se omogućila ocena rezultata, može se računati foto-iritacioni faktor (Photo-Irritation-Factor, PIF) ili srednji foto-efekat (Mean Photo Effect, MPE).
Za izračunavanje mera fototoksičnosti (videti ispod) potrebno je aproksimovati skup pojedinačnih vrednosti koncentracija-odgovor pomoću odgovarajuće neprekidne (model) krive koncentracija - odgovor. Podešavanje krive prema podacima obično se izvodi ne-linearnom regresivnom metodom18. Da bi se procenio uticaj varijabilnosti podataka na podešavanje krive, preporučuje se postupak bootstrap.
Foto-iritacioni faktor (u daljem tekstu: PIF) izračunava se upotrebom formule:
PIF= | IC50(-Irr) | |
IC50(+Irr) |
Ako se IC50 u prisustvu i u odsustvu svetlosti ne može izračunati, PIF se ne može odrediti za ispitivani materijal. Srednji foto-efekat (u daljem tekstu: MPE) određuje se na osnovu upoređivanja završenih koncentracija-odgovor krivih.
Foto-efekat PEc pri bilo kojoj koncentraciji C se definiše kao proizvod odgovora efekta REc i doze efekta DEc tj. PEc = REc x DEc. Odgovor efekta REc je razlika između odgovora primećenog u odsustvu i u prisustvu svetlosti, tj. REc = Rc (-Irr) − Rc (+Irr). Doza-efekat se daje kao:
DEc= | C/C * - 1 | |
C/C * + 1 |
Pri čemu C* jeste ravnotežna koncentracija, odnosno koncentracija pri kojoj je +Irr odgovor jednak -Irr odgovoru pri koncentraciji C. Ako se C* ne može odrediti zato što su vrednosti odgovora +Irr krive sistematski više ili niže nego RC(-Irr), dozni efekat je 1. Faktori wi su dati po najvišoj vrednosti odgovora, tj. wi = MAX {Ri (+Irr), Ri (-Irr)}. Koncentracija Ci se bira tako da isti broj tačaka pripadne svakom koncentracionom intervalu koji su definisani sa vrednosti koncentracije pri kojoj najmanje jedna ili dve krive i dalje pokazuju vrednost odgovora od najmanje 10%. Ako je ova maksimalna koncentracija viša od najviše koncentracije koja je korišćena u +Irr eksperimentu, onda se rezidualni deo +Irr krive podešava da je vrednost odgovora "0". U zavisnosti od toga da li je MPE vrednost veća od određene cut-off vrednosti (MPEc = 0,15) ili nije, hemikalija se klasifikuje kao fototoksična.
Softverski paket za izračunavanje PIF i MPE je dostupan20.
Na osnovu studije validacije8, ispitivana supstanca sa PIF < 2 ili sa MPE < 0,1 predviđa: "nije fototoksično". Ako je PIF > 2 i < 5 ili ako je MPE > 0,1 i < 0,15 predviđa: "verovatna fototoskičnost"; i ako je PIF > 5 ili MPE > 0,15 predviđa: "fototoksičnost".
Za laboratorije koje uvode ovaj test, treba ispitati referentne materijale nabrojane u Tabeli 1. pre ispitivanja ispitivane supstance za procenu fototoksičnosti. Vrednosti PIF i MPE treba da budu približne vrednostima pomenutim u Tabeli 1.
Tabela 1.
Hemijski naziv | EINECS broj | CAS broj | PIF | MPE | Apsorpcioni maksimum | RastvaračLII |
amiodaron HCL | 243-293-2 | [19774-82-4] | > 3,25 | 0,2-0,54 | 242 nm 300 nm | etanol |
hlorpromazin HCL | 200-701-3 | [69-09-0] | > 14,4 | 0,33-0,63 | 309 nm | etanol |
norfloksacin | 274-614-4 | [70458-96-7] | > 71,6 | 0,34-0,90 | 316 nm | acetonitril |
antracen | 204-371-1 | [120-12-7] | > 18,5 | 0,19-0,81 | 356 nm | acetonitril |
Protoporfirin IX, dinatrijum | 256-815-9 | [50865-01-5] | > 45,3 | 0,54-0,74 | 402 nm | etanol |
L-histidin |
| [7006-35-1] |
| 0,05-0,10 | 211 nm | voda |
heksahlorofen | 200-733-8 | [70-30-4] | 1,1-1,7 | 0,00-0,05 | 299 nm 317 nm | etanol |
Natrijum-lauril sulfat | 205-788-1 | [151-21-3] | 1,0-1,9 | 0,00-0,05 | Nema apsorpcije | voda |
______________
LII Rastvarač koji se koristi za merenje apsorbancije.
Fototoksični efekti se uočavaju samo pri najvišim ispitivanim koncentracijama (naročito za ispitivane hemikalije koje su rastvorne u vodi). Dodatna razmatranja mogu biti neophodna za procenu hazarda. Ovo može uključiti podatke o apsorpciji kože i o akumulaciji hemikalije u koži i/ili podatke iz drugih testova, npr. ispitivanje hemikalije na in vitro životinjskoj ili ljudskoj koži ili modelima kože.
Ako nije pokazana toksičnost (+Irr i -Irr) i ako je slaba rastvorljivost bila ograničavajući faktor za koncentracije koje mogu biti ispitane, može se dovesti u pitanje kompatibilnost ispitivane supstance sa testom i treba razmotriti potvrdno testiranje upotrebom npr. drugog modela.
Izveštaj o ispitivanju mora da sadrži podatke o:
1) Ispitivanoj supstanci:
- podaci o identifikaciji, uobičajeni generički nazivi i IUPAC i CAS brojevi, ako su poznati;
- fizička priroda i čistoća;
- fizičke i hemijske osobine relevantne za izvođenje studije;
- UV/vis apsorpcioni spektar;
- stabilnost i fotostabilnost, ako je poznata.
2) Rastvaraču:
- opravdanje za izbor rastvarača;
- rastvorljivost ispitivane hemikalije u rastvaraču;
- procenat prisustva rastvarača u medijumu za tretman.
3) Ćelijama:
- vrsta i izvor ćelija;
- odsustvo mikoplazme;
- broj ćelijskih prolaza, ako je poznat;
- osetljivost ćelija na zračenje, određena opremom za zračenje koja se koristi u in vitro 3T3 NRU testu fototoksičnosti.
4) Uslovima ispitivanja1; inkubaciji pre i posle tretmana:
- vrsta i sastav medijuma za kultivisanje;
- uslovi inkubacije (koncentracija CO2; temperatura; vlažnost);
- trajanje inkubacije (predtretman, post-tretman).
5) Uslovima ispitivanja2; tretmanu hemikalijom:
- osnova za izbor koncentracija ispitivane hemikalije koje su korišćene u prisustvu i u odsustvu zračenja;
- u slučaju ograničene rastvorljivosti ispitivane hemikalije i odsustva citotoksičnosti: osnova za najvišu ispitivanu koncentraciju;
- vrsta sastava medijuma za tretiranje (puferovani rastvor soli);
- trajanje hemijskog tretmana.
6) Uslovima ispitivanja3; zračenju:
- osnova za izbor izvora svetlosti koji je korišćen;
- proizvođač i vrsta izvora svetlosti i radiometra;
- karakteristike spektralnog zračenja izvora svetlosti;
- transmisione i apsorpcione karakteristike korišćenih filtera;
- karakteristike radiometra i pojedinosti o kalibraciji;
- razmak svetlosnog izvora od ispitivanog sistema;
- UVA zračenje pri ovom razmaku, izraženo u mW/cm2;
- trajanje izlaganja UV/vis svetlosti;
- UVA doza (zračenje x vreme), izražena u J/cm2;
- temperatura ćelijskih kultura tokom zračenja i ćelijskih kultura koje se istovremeno drže u mraku.
7) Uslovima ispitivanja4; Neutral Red testu viabilnosti:
- sastav Neutral Red medijuma za tretiranje;
- trajanje inkubacije sa Neutral Red;
- uslovi inkubacije (koncentracija CO2; temperatura; vlažnost);
- uslovi ekstrakcije Neutral Red (ekstrakcija; trajanje);
- talasna dužina koja je korišćena za spektrofotometrijsko čitanje optičke gustine Neutral Red;
- druga talasna dužina (referentna), ako je korišćena;
- sadržaj slepe probe spektrofotometra, ako je korišćena.
8) Rezultatima:
- viabilnost ćelija dobijena iz svake koncentracije ispitivane hemikalije, izražena u procentu viabilnosti od srednje vrednosti istovremene kontrole rastvarača;
- krive koncentracija-odgovor (koncentracija ispitivane supstance prema relativnoj viabilnosti ćelija) dobijene u istovremenim +Irr i -Irr ispitivanjima;
- analiza krivih koncentracija-odgovor: ako je moguće, izračunavanje IC50 (+Irr) i IC50 (-Irr);
- upoređivanje dve krive koncentracija-odgovor dobijene u prisustvu i u odsustvu zračenja, bilo računanjem foto-iritacionog faktora (PIF), bilo računanjem srednjeg foto-efekta (MPE);
- kriterijumi za prihvatanje testa; istovremena kontrola rastvarača;
- apsolutna viabilnost (optička gustina ekstrakta Neutral Red) ozračenih i neozračenih ćelija;
- istorijski podaci negativne i kontrole rastvarača; srednje vrednosti i standardne devijacije;
- kriterijumi za prihvatanje testa; istovremena pozitivna kontrola;
- IC50(+Irr) i IC50(-Irr) i PIF/MPE hemikalije koja je korišćena kao pozitivna kontrola;
- istorijski podaci o hemikaliji koja je korišćena kao pozitivna kontrola: IC50(+Irr) i IC50(-Irr) i PIF/MPE; srednje vrednosti i standardne devijacije.
9) Obrazloženju rezultata.
10) Zaključcima.
1. Lovell W.W., (1993) A scheme for in vitro screening of substances for photoallergenic potential. Toxicology In vitro 7, p. 95-102.
2. Santamaria, L. and Prino, G., (1972) List of the photodynamic substances. In "Research Progress in Organic, Biological and Medicinal Chemistry" Vol. 3 part 1. North Holland Publishing Co. Amsterdam. p. XI-XXXV.
3. Spielmann, H., Lovell, W.W., Hölzle, E., Johnson, B.E., Maurer, T., Miranda, M.A., Pape, W.J.W., Sapora, O., and Sladowski, D., (1994) In vitro phototoxicity testing: The report and recommendations of ECVAM Workshop 2. ATLA, 22, p. 314-348.
4. Spikes, J.D., (1989) Photosensitisation. In "The science of Photobiology" Edited by K.C. Smith. Plenum Press, New York. 2nd edition, p. 79-110.
5. OECD, (1997) Environmental Health and Safety Publications, Series on Testing and Assessment No 7 "Guidance Document On Direct Phototransformation Of Chemicals In Water" Environment Directorate, OECD, Paris.
6. Spielmann, H., Balls, M., Döring, B., Holzhütter, H.G., Kalweit, S., Klecak, G., L'Eplattenier, H., Liebsch, M., Lovell, W.W., Maurer, T., Moldenhauer. F. Moore. L., Pape, W., Pfannbecker, U., Potthast, J., De Silva, O., Steiling, W., and Willshaw, A., (1994) EEC/COLIPA project on in vitro phototoxicity testing: First results obtained with a Balb/c 3T3 cell phototoxicity assay. Toxic. In vitro 8, p. 793-796.
7. Anon, (1998) Statement on the scientific validity of the 3T3 NRU PT test (an in vitro test for phototoxicity), European Commission, Joint Research Centre: ECVAM and DGXI/E/2, 3 November 1997, ATLA, 26, p. 7-8.
8. Spielmann, H., Balls, M., Dupuis, J., Pape, W.J.W., Pechovitch, G. De Silva, O., Holzhütter, H.G., Clothier, R., Desolle, P., Gerberick, F., Liebsch, M., Lovell, W.W., Maurer, T., Pfannenbecker, U., Potthast, J. M., Csato, M., Sladowski, D., Steiling, W., and Brantom, P., (1998) The international EU/COLIPA In vitro phototoxicity validation study: results of phase II (blind trial), part 1: the 3T3 NRU phototoxicity test. Toxicology In vitro 12, p. 305-327.
9. OECD, (2002) Extended Expert Consultation Meeting on The In vitro 3T3 NRU Phototoxicity Test Guideline Proposal, Berlin, 30th-31th October 2001, Secretariat's Final Summary Report, 15th March 2002, OECD ENV/EHS, available upon request from the Secretariat.
10. Borenfreund, E., and Puerner, J.A., (1985) Toxicity determination in vitro by morphological alterations and neutral red absorption. Toxicology Lett., 24, p. 119-124.
11. Hay, R.J., (1988) The seed stock concept and quality control for cell lines. Analytical Biochemistry 171, p. 225-237.
12. Lambert L.A, Warner W.G., and Kornhauser A., (1996) Animal models for phototoxicity testing. In 'Dermatotoxicology', edited by F.N. Marzulli and H.I. Maibach. Taylor & Francis, Washington DC. 5th Edition, p. 515-530.
13. Tyrrell R.M., Pidoux M., (1987) Action spectra for human skin cells: estimates of the relative cytotoxicity of the middle ultraviolet, near ultraviolet and violet regions of sunlight on epidermal keratinocytes. Cancer Res., 47, p. 1825-1829.
14. ISO 10977., (1993) Photography - Processed photographic colour films and paper prints - Methods for measuring image stability.
15. Sunscreen Testing (UV.B) TECHNICALREPORT, CIE, International Commission on Illumnation, Publication No 90, Vienna, 1993, ISBN 3 900 734 275
16. ZEBET/ECVAM/COLIPA - Standard Operating Procedure: In vitro 3T3 NRU Phototoxicity Test. Final Version, 7 September, 1998. p. 18.
17. Spielmann, H., Balls, M., Dupuis, J., Pape, W.J.W., De Silva, O., Holzhütter, H.G., Gerberick, F., Liebsch, M., Lovell, W.W., and Pfannenbecker, U., (1998) A study on UV filter chemicals from Annex VII of the European Union Directive 76/768/EEC, in the in vitro 3T3 NRU phototoxicity test. ATLA 26, p. 679-708.
18. Holzhütter, H.G., and Quedenau, J., (1995) Mathematical modeling of cellular responses to external signals. J. Biol. Systems 3, p. 127-138.
19. Holzhütter, H.G., (1997) A general measure of in vitro phototoxicity derived from pairs of dose-response curves and its use for predicting the in vivo phototoxicity of chemicals. ATLA, 25, p. 445-462.
20. http://www.oecd.org/document/55/0,2340,en_2649_34377_2349687_1_1_1_1,00.html
ULOGA 3T3 NRU PT U PRISTUPU KORAK PO KORAK ZA ISPITIVANJE FOTOTOKSIČNOSTI HEMIKALIJA
SLIKA 1: RASPODELA SPEKTRALNE MOĆI FILTRIRANOG SOLARNOG SIMULATORA
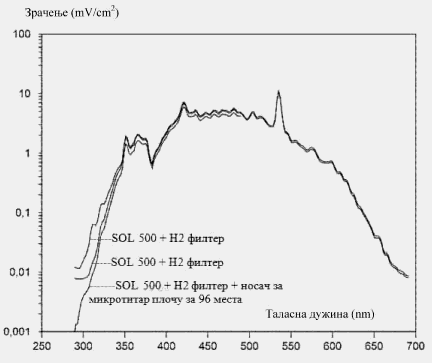
(videti odeljak 1.4.1.5, drugi pasus ove metode)
Slika 1. daje primer prihvatljive raspodele spektralnog zračenja filtriranog solarnog simulatora. On je od punjenog metal halofenog izvora koji je korišćen u probi validacije 3T3 NRU PT6, 8, 17. Prikazani su efekat dva različita filtera i dodatni efekat filtriranja poklopca posude sa 96 bazenčića. Filter H2 je korišćen samo sa test sistemima koji mogu da tolerišu veću količinu UVB (test modela kože i test foto-hemolize eritrocita). U 3T3 NRU PT korišćen je H1 filter. Slika pokazuje da je dodatni efekat filtriranja poklopca posude uglavnom uočen u opsegu UVB, ali i dalje ostavljajući dovoljno UVB spektar zračenja da se pobude hemikalije koje tipično apsorbuju UVB opseg, kao što je amjodaron (videti Tabelu 1.).
Slika 2: Osetljivost na zračenje Balb/c 3T3 ćelija (izmereno u opsegu UVA)
Viabilnost ćelija (% preuzimanja Neutral Red od strane kontrola u mraku)
(videti odeljak 1.4.1.5.2. drugi pasus i odeljke 1.4.2.2.1. i 1.4.2.2.2. ove metode)
Osetljivost Balb/c 3T3 ćelija na zračenje sa solarnim simulatorom koji je korišćen u probi validacije 3T3 NRU testa fototoksičnosti, merena u UVA opsegu. Slika pokazuje rezultate dobijene u sedam različitih laboratorija u prevalidacionoj studiji1. Zbog toga što su obe krive sa otvorenim znakovima dobijene sa ostarelim ćelijama (veliki broj prolaza), one su morale da budu zamenjene novim sa zadebljanim oznakama koje pokazuju prihvatljivu toleranciju na zračenje, koje su dobijene na novim ćelijama. Iz ovih podataka izvedena je najviša doza zračenja koja nije toksična, od 5 J/cm2. Horizontalna isprekidana linija dodatno pokazuje maksimalno prihvatljiv efekat zračenja dat u odeljku 1.4.2.2. ove metode.
B.42. SENZIBILIZACIJA KOŽE: TEST LOKALNIH LIMFNIH ČVOROVA
Ova eksperimentalna metoda potpuno odgovara metodi OECD: TG 429 (2002).
Test lokalnih limfnih čvorova (Local Lymph Node Assay, u daljem tekstu: LLNA) je u dovoljnoj meri proveren i prihvaćen da se opravda njegovo prihvatanje kao nove metode1, 2, 3. Ovo je druga metoda za ocenjivanje potencijala hemikalije da izazove senzibilizaciju kože kod životinja. Druga metoda (V.6. koja je data u ovom prilogu) koristi zamorce, pre svega test maksimizacije i Buehlerov test4.
LLNA daje alternativnu metodu za identifikaciju hemikalija koje izazivaju senzibilizaciju kože i za potvrđivanje da hemikalije nemaju značajan potencijal da uzrokuju senzibilizaciju kože. Ovo ne znači da LLNA treba uvek koristiti umesto ispitivanja na zamorcima, već pre da je ovaj test jednako dobar i da može biti primenjen kao alternativa u kojoj pozitivni i negativni rezultati uglavnom ne zahtevaju dalju potvrdu.
LLNA pruža određene prednosti u odnosu na naučni napredak i dobrobit životinja. Test proučava fazu indukcije senzibilizacije kože i daje kvantitativne podatke pogodne za procenu odnosa doza-odgovor. Detalji validacije LLNA i pregled rada u vezi s tim su objavljeni5, 6, 7, 8. Treba imati u vidu da su blagi/umereni senzibilizatori koji su preporučeni kao pogodne supstance za pozitivnu kontrolu za testove na zamorcima, takođe pogodni za korišćenje u LLNA6, 8, 9.
LLNA je in vivo metoda i ne eliminiše korišćenje životinja u ocenjivanju aktivnosti kontaktne senzibilizacije. Ona ima potencijal da smanji broj životinja koji je potreban za ovu svrhu. LLNA nudi značajno poboljšanje u načinu na koji se životinje koriste za testiranje kontaktne senzibilizacije. LLNA se zasniva na razmatranju imunoloških pojava koje su stimulisane hemikalijama za vreme indukcione faze senzibilizacije. Za razliku od ispitivanja na zamorcima, LLNA ne zahteva primenu osporavanih dermalnih reakcija preosetljivosti. Ova metoda ne zahteva korišćenje adjuvansa, kao što je slučaj kod testa maksimizacije na zamorcima. Zbog toga LLNA smanjuje stres kod životinja. Uprkos prednostima ove metode u odnosu na tradicionalne testove na zamorcima, postoje određena ograničenja koja mogu zahtevati tradicionalna ispitivanja na zamorcima (npr. lažno negativni rezultati LLNA sa nekim metalima, lažno pozitivni rezultati sa nekim iritansima kože)10.
Videti Opšti uvod.
Osnovni princip u LLNA je da senzibilišuće supstance dovode do primarne proliferacije limfocita u limfnom čvoru koji drenira mesto primene hemikalije. Ova proliferacija je proporcionalna primenjenoj dozi (i potenciji alergena) i predstavlja jednostavno sredstvo za dobijanje objektivne, kvantitativne mere senzibilizacije. LLNA procenjuje ovu proliferaciju kao odnos doza-odgovor u kojem se proliferacija u ispitivanim grupama upoređuje sa onom kod kontrola tretiranih rastvaračem. Odnos proliferacije u tretiranim grupama prema onima u kontrolama rastvarača, nazvana "indeks stimulacije", određuje se i mora da bude najmanje tri, pre nego što se ispitivana supstanca dalje ocenjuje kao potencijalno senzibilišuća za kožu. Ovde opisane metode zasnivaju se na korišćenju radioaktivnog obeležavanja kako bi se izmerila proliferacija ćelija. Drugi pokazatelji ispitivanja za ocenjivanje proliferacije mogu se primeniti ukoliko postoji opravdanje i odgovarajuća naučna podrška, uključujući potpune navode i opis metodologije.
1.3.1. Pripreme
1.3.1.1. Uslovi smeštaja i ishrane
Životinje treba da budu smeštene pojedinačno. Temperatura prostorija za eksperimentalne životinje treba da bude 22°C (±3°C). Relativna vlažnost treba da bude najmanje 30% i da ne prelazi 70% osim za vreme čišćenja prostorija. Ciljna vlažnost treba da bude 50% do 60%. Svetlo treba da bude veštačko sa sekvencama od 12 sati svetla i 12 sati mraka. Može da se koristi konvencionalna laboratorijska ishrana uz slobodan pristup vodi za piće.
1.3.1.2. Priprema životinja
Životinje se biraju nasumce i označavaju da bi se omogućila identifikacija jedinki (ali ne označavanjem ušiju na bilo koji način) i drže se u kavezima najmanje pet dana pre početka doziranja kako bi se aklimatizovale na laboratorijske uslove. Pre početka tretmana sve životinje se pregledaju da se utvrdi da nemaju vidljivih lezija na koži.
1.3.2. Uslovi ispitivanja
1.3.2.1. Eksperimentalne životinje
Za ovaj test koristi se miš. Ispitivanje se izvodi na mladim odraslim ženskama miševa soja CBA/Ca ili CBA/J, koje nisu ranije rađale i nisu gravidne. Na početku studije životinje treba da budu između 8 i 12 nedelja stare i varijacija u telesnoj masi životinja treba da bude minimalna i ne sme da prelazi 20% prosečne telesne mase. Drugi sojevi i mužjaci mogu se koristiti kada se prikupi dovoljno podataka da se pokaže da ne postoje značajne razlike u LLNA odgovoru između određenih sojeva i/ili polova.
1.3.2.2. Provera pouzdanosti
Pozitivne kontrole se koriste kako bi se pokazale odgovarajuće osobine testa i kompetentnost laboratorije da uspešno obavi studiju. Pozitivna kontrola treba da dovede do pozitivnog LLNA odgovora pri nivou ekspozicije za koji se očekuje da da povećanje indeksa stimulacije (SI) >3 u odnosu na negativnu kontrolnu grupu. Doza pozitivne kontrole treba da bude odabrana tako da indukcija bude jasna ali ne prekomerna. Poželjno je primeniti supstance: heksil cinamin aldehid (CAS broj 101-86-0, EINECS broj 202-983-3) i merkaptobenzotiazol (CAS broj 149-30-4, EINECS broj 205-736-8). Mogu postojati okolnosti u kojima se, uz dovoljno opravdanje, mogu koristiti druge kontrolne supstance koje zadovoljavaju gore navedene kriterijume. Grupa pozitivne kontrole može biti potrebna u svakom testu, mogu postojati situacije u kojima će laboratorije imati istorijske podatke pozitivne kontrole koji pokazuju konzistentnost zadovoljavajućeg efekta tokom šestomesečnog ili dužeg perioda. U tim slučajevima može biti pogodno ređe testiranje s pozitivnim kontrolama u periodima kraćim od 6 meseci. Mada pozitivna kontrolna supstanca treba da bude ispitana u vehikulumu za koje je poznato da izaziva konzistentan odgovor (npr. aceton: maslinovo ulje), može doći do nekih propisanih situacija u kojima će biti potrebno testiranje nestandardnog vehikuluma (klinički/hemijski relevantna formulacija). U takvim situacijama treba ispitati moguću interakciju pozitivne kontrole sa ovim nekonvencionalnim vehikulumom.
1.3.2.3. Broj životinja, dozni nivoi i izbor vehikuluma.
Ispitivanje se vrši na najmanje četiri životinje po doznoj grupi, sa najmanje tri koncentracije ispitivane supstance, plus grupa negativne kontrole koja se tretira samo vehikulumom za ispitivanu supstancu i, ako odgovara, pozitivna kontrola. U slučajevima u kojima treba prikupiti podatke o pojedinačnim životinjama, koristi se najmanje pet životinja po doznoj grupi. Osim u slučaju odsustva tretmana ispitivanom supstancom, sa životinjama u kontrolnoj grupi treba postupati na isti način kao i sa životinjama u tretiranim grupama.
Izbor doze i vehikuluma treba da bude zasnovan na preporukama iz literaure1. Doze se biraju iz niza koncentracija: 100%, 50%, 25%, 10%, 5%, 2.5%, 1%, 0.5%, itd. Kada su dostupni, treba razmotriti postojeće podatke o akutnoj toksičnosti i iritaciji kože prilikom izbora tri uzastopne koncentracije tako da najveća koncentracija maksimizuje izlaganje uz izbegavanje sistemske toksičnosti i prekomerne lokalne iritacije kože2, 11.
Vehikulum se bira na osnovu maksimizacije ispitivanih koncentracija i rastvorljivosti uz pravljenje rastvora/suspenzije koja odgovara za primenu ispitivane supstance. Preporučeni vehikulumi (navedeni od više ka manje odgovarajućim) su aceton/maslinovo ulje (4:1 v/v), dimetilformamid, metil etil keton, propilen glikol i dimetil sulfoksid2, 10, ali se mogu koristiti i drugi uz zadovoljavajuće naučno obrazloženje. U određenim situacijama može da bude neophodno da se koristi klinički relevantni rastvarač ili komercijalna formulacija u kojoj se ispitivana supstanca stavlja na tržište kao dodatna kontrola. Posebnu pažnju treba posvetiti da se obezbedi da se hidrofilni materijali inkorporiraju u sistem vehikuluma, koji vlaži kožu i ne skida se trenutno. Izbegavaju se potpuno vodeni vehikulumi.
1.3.3. Postupak ispitivanja
1.3.3.1. Eksperimentalni raspored
Eksperimentalni raspored studije:
Dan 1:
Pojedinačno se identifikuju životinje i beleži se telesna masa svake životinje. Započinje se aplikaciju 25 µl odgovarajućeg rastvora ispitivane supstance, samog vehikuluma ili pozitivne kontrole (kako je odgovarajuće) na poleđinu svakog uha.
Dani 2 i 3:
Ponavlja se postupak aplikacije koji je primenjen dana 1.
Dani 4 i 5:
Bez tretmana.
Dan 6:
Beleži se telesna masa svake životinje. Ubrizgava se 250 µl rastvora soli sa fosfatnim puferom (u daljem tekstu: PBS) koja sadrži 20 µCi (7.4e + 8 Bq) 3H -metil timidina svim ispitivanim i kontrolnim miševima putem repne vene. Alternativno se ubrizgava 250 µl PBS koji sadrži 2 µCi (7.4e + 7 Bq) 125I jododeoksiuridina i 10-5 M fluorodeoksiuridina svim miševima putem repne vene.
Posle pet sati životinje se žrtvuju. Iseku se ušni drenažni limfni čvorovi svakog uha i sakupljaju u PBS za svaku eksperimentalnu grupu (pristup zbirne tretirane grupe). Alternativno se mogu iseći parovi limfnih čvorova pojedinačnih životinja i staviti u PBS za svaku životinju (individualni pristup). Detalji i dijagrami identifikacije čvorova i disekcije dati su u literaturi10.
1.3.3.2. Priprema suspenzija ćelija
Jedna suspenzija ćelija limfnih čvorova (u daljem tekstu: LNC) od zbirnih tretiranih grupa ili obostrano od pojedinačnih životinja priprema se nežnim mehaničkim razdavanjem kroz gazu od nerđajućeg čelika mrežice 200 µm. Ćelije limfnog čvora dva puta se ispiraju s preostalim PBS i talože sa 5% trihlorsirćetnom kiselinom (u daljem tekstu: TCA) na 4 °C tokom 18 sati2. Zrnca se ponovno suspenduju u 1 ml TCA i prenose u scintilacijske bočice koje sadrže 10 ml scintilacione tečnosti za 3H-brojanje ili se direktno prenose u epruvete za gama brojenje za 125I-prebrojavanje.
1.3.3.3. Određivanje proliferacije ćelija (inkorporirana radioaktivnost)
Inkorporiranje 3H-metil timidina se meri β-scintilacionim brojanjem kao raspadanje po minuti (u daljem tekstu: DPM). Inkorporiranje 125I-jododeoksiuridina se meri 125I-brojanjem i izražava kao DPM. Zavisno od pristupa, dodavanje se izražava kao DPM/tretirana grupa (pristup zbirne tretirane grupe) ili DPM/životinja (individualni pristup).
1.3.3.4. Zapažanja
1.3.3.4.1. Klinička zapažanja
Životinje treba pažljivo pregledati jednom dnevno zbog kliničkih znakova, bilo lokalne iritacije na mestu aplikacije ili zbog sistemske toksičnosti. Sva zapažanja se sistematično beleže u pojedinačnim dokumentima koji se vode za svaku životinju.
1.3.3.4.2. Telesna masa
Kako je navedeno u odeljku 1.3.3.1. ove metode, individualne telesne mase životinja treba izmeriti na početku ispitivanja i prilikom planiranog žrtvovanja životinja.
1.3.4. Izračunavanje rezultata
Rezultati su izraženi kao indeks stimulacije (u daljem tekstu: SI). Kada se koristi pristup zbirne tretirane grupe, SI se dobija deljenjem zbirne radioaktivne inkorporacije za svaku tretiranu grupu sa inkorporacijom zbirne grupe kontrole vehikuluma. Na ovaj način se dobija srednji SI. Prilikom korišćenja individualnog pristupa, SI se dobija deljenjem srednje DPM/životinja unutar svake grupe ispitivane supstance i grupe pozitivne kontrole srednjom DPM/životinja za kontrolnu grupu rastvarača/vehikuluma. Srednji SI za kontrole tretirane vehikulumom je tada 1.
Korišćenje individualnog pristupa za izračunavanje SI omogućava statističku analizu podataka. Prilikom izbora odgovarajuće metode statističke analize istraživač treba da vodi računa o mogućim nejednakostima varijansi i drugim relevantnim problemima koji mogu da zahtevaju transformaciju podataka ili neparametarske statističke analize. Adekvatni pristup za interpretaciju podataka je da se ocene svi individualni podaci tretiranih i kontrola vehikuluma i da se iz njih izvede najbolja kriva doza-odgovor, uzimajući u obzir granice pouzdanosti8, 12, 13. Istraživač treba da vodi računa o mogućim "nepodobnim" reakcijama kod pojedinih životinja unutar grupe koje mogu zahtevati korišćenje alternativne mere odgovora (npr. mediana, a ne srednja vrednost) ili eliminaciju nepodobnih rezultata.
Proces donošenja odluka u pogledu pozitivnog odgovora uključuje indeks stimulacije ≥ 3 zajedno sa razmatranjem odnosa doza-odgovor i, kada odgovara, statistički značaj3, 6, 8, 12, 14.
Ukoliko je neophodno razjasniti dobijene rezultate, treba razmotriti razna svojstva ispitivane supstance, uključujući i to da li ima strukturne sličnosti sa supstancama za koje je poznato da su senzibilišuće za kožu, da li uzrokuje prekomernu iritaciju kože i prirodu opaženog odnosa doze i odgovora. Ova i druga pitanja detaljno su razmatrana na drugom mestu7.
Podaci se navode tabelarno. Navode se srednje i pojedinačne DPM vrednosti i indeksi stimulacije za svaku doznu grupu (uključujući kontrolu vehikuluma).
Izveštaj o ispitivanju, ukoliko je moguće, sadrži podatke o:
1) Ispitivanoj supstanci:
- identifikacioni podaci (npr. CAS broj, ukoliko je dostupan; izvor; čistoću; poznate nečistoće; broj serije);
- fizičku prirodu i fizička i hemijska svojstva (npr. isparljivost, stabilnost, rastvorljivost);
- u slučaju smeše, sastav i relativna zastupljenost (procenat) sastojaka.
2) Vehikulumu:
- identifikacioni podaci (čistoća; koncentracija, gde odgovara; korišćena zapremina);
- opravdanje za izbor vehikuluma.
3) Eksperimentalnim životinjama:
- soj korišćenih miševa;
- mikrobiološki status životinja, kada je poznat;
- broj, starost i pol životinja;
- izvor životinja, uslovi smeštaja, ishrana, itd.
4) Uslovima ispitivanja:
- detalji o pripremi ispitivane supstance i primeni;
- opravdanje za izbor doze, uključujući rezultate studije raspona, ukoliko je obavljena; korišćen vehikulum i koncentracije ispitivne supstance i ukupna količina primenjene supstance;
- detalji o kvalitetu hrane i vode (uključujući vrstu ishrane/izvor, izvor vode).
5) Kontroli pouzdanosti:
- sažetak rezultata zadnje provere pouzdanosti uključujući podatke o supstanci, koncentraciji i korišćenom vehikulumu;
- podaci o istovremenoj i/ili istorijskoj pozitivnoj i negativnoj kontroli za laboratoriju koja vrši ispitivanje.
6) Rezultatima:
- pojedinačna telesna masa životinja na početku doziranja i u vreme planiranog žrtvovanja;
- tabela srednjih (pristup zbirne tretirane grupe) i individualnih (individualni pristup) DPM vrednosti kao i raspon vrednosti za oba pristupa i stimulacioni indeksi za svaku doznu grupu (uključujući kontrolu vehikuluma);
- statistička analiza, kad to odgovara;
- vreme pojave i znaci toksičnosti, uključujući iritaciju kože na mestu primene, ukoliko ih ima, za svaku životinju.
7) Obrazloženju rezultata:
- kratak komentar rezultata, analiza odnosa doza-odgovor i statističke analize, kada odgovara, sa zaključkom da li treba da se ispitivana supstanca smatra senzibilišućom za kožu.
1. Kimber, I. and Basketter, D.A. (1992). The murine local lymph node assay; collaborative studies and new directions: A commentary. Food and Chemical Toxicology 30, 165-169.
2. Kimber, I, Derman, R.J., Scholes E.W, and Basketter, D.A. (1994). The local lymph node assay: developments and applications. Toxicology, 93, 13-31.
3. Kimber, I., Hilton, J., Dearman, R.J., Gerberick, G.F., Ryan, C.A., Basketter, D.A., Lea, L., House, R.V., Ladies, G.S., Loveless, S.E., Hastings, K.L. (1998). Assessment of the skin sensitisation potential of topical medicaments using the local lymph node assay: An interlaboratory exercise. Journal of Toxicology and Environmental Health, 53, 563-79.
4. Testing Method B.6.
5. Chamberlain, M. and Basketter, D.A. (1996). The local lymph node assay: status of validation. Food and Chemical Toxicology, 34, 999-1002.
6. Basketter, D.A., Gerberick, G.F., Kimber, I. and Loveless, S.E (1996). The local lymph node assay- A viable alternative to currently accepted skin sensitisation tests. Food and Chemical Toxicology, 34, 985- 997.
7. Basketter, D.A., Gerberick, G.F. and Kimber, I. (1998). Strategies for identifying false positive responses in predictive sensitisation tests. Food and Chemical Toxicology. 36, 327-33.
8. Van Och, F.M.M, Slob, W., De Jong, W.H., Vandebriel, R.J., Van Loveren, H. (2000). A quantitative method for assessing the sensitising potency of low molecular weight chemicals using a local lymph node assay: employement of a regression method that includes determination of uncertainty margins. Toxicology, 146, 49-59.
9. Dearman, R.J., Hilton, J., Evans, P., Harvey, P., Basketter, D.A. and Kimber, I. (1998). Temporal stability of local lymph node assay responses to hexyl cinnamic aldehyde. Journal of Applied Toxicology, 18, 281-4.
10. National Institute of Environmental Health Sciences (1999). The Murine Local Lymph Node Assay: A Test Method for Assessing the Allergic Contact Dermatitis Potential of Chemicals/Compounds: The Results of an Independent Peer Review Evaluation Coordinated by the Interagency Coordinating Committee on the Validation of Alternative Methods (ICCVAM) and the National Toxicology Program Center for the Evaluation of Alternative Toxicological Methods (NICETAM). NIH Publication No: 99-4494, Research Triangle Park, N.C. (http://iccvam.niehs.nih.gov).
11. Testing method B.4.
12. Basketter, D.A., Selbie, E., Scholes, E.W. Lees, D. Kimber, I. and Botham, P.A. (1993) Results with OECD recommended positive control podraživače in the maximisation, Buehler and local lymph node assays. Food and Chemical Toxicology, 31, 63-67.
13. Basketter D.A., Lea L.J., Dickens A., Briggs D., Pate I., Dearman R.J., Kimber I. (1999). A comparison of statistical approaches to the derivation of EC3 values from local lymph node assay dose responses. J. Appl. Toxicology, 19, 261-266.
14. Basketter DA, Blaikie L, Derman RJ, Kimber I, Ryan CA, Gerberick GF, Harvey P, Evans P, White IR and Rycroft RTG (2000). Use of local lymph node assay for the estimation of relative contact allergenic potency. Contact Dermatitis 42, 344-48.
B.43. STUDIJA NEUROTOKSIČNOSTI KOD GLODARA
Ova metoda potpuno odgovara metodi OECD: TG 424 (1997).
Ova metoda ispitivanja je osmišljena za dobijanje podataka potrebnih za potvrdu ili dalju karakterizaciju potencijalne neurotoksičnosti hemikalija kod odraslih životinja. Može se kombinovati sa postojećim metodama ispitivanja za studije toksičnosti ponovljene doze ili obavljati kao posebna studija. Preporučuje se konsultacija OECD vodiča1 oko pomoći pri osmišljavanju studije zasnovane na ovoj metodi ispitivanja. Navedeno je posebno važno kod razmatranja modifikacija opažanja i postupaka ispitivanja kao što se preporučuje za rutinsku primenu ove metode. Pripremljen je dokument sa smernicama kako bi omogućio izbor drugih postupaka ispitivanja za upotrebu u posebnim okolnostima.
Procena razvojne neurotoksičnosti nije predmet ove metode.
U proceni i evaluaciji toksičnih karakteristika hemikalija važno je uzeti u obzir potencijal za neurotoksične efekte. Metoda za sistemsku toksičnost, ponovljena doza već uključuje posmatranja koja se odnose na potencijalnu neurotoksičnost. Ova metoda ispitivanja može se koristiti pri osmišljavanju studije za dobijanje daljih podataka o ili za potvrdu neurotoksičnih efekata opaženih u studijama za sistemske toksičnost, ponovljena doza. Razmatranje potencijalne neurotoksičnosti određenih grupa hemikalija može ukazati na to da se one mogu odgovarajuće oceniti ovom metodom bez prethodnih indikacija potencijalne neurotoksičnosti iz studija sistemske toksičnosti, ponovljena doza. Takva razmatranja npr. uključuju:
- posmatranje neuroloških znakova ili neuropatoloških lezija u studijama toksičnosti različitih od onih iz studija sistemsku toksičnost, ponovljena doza ili
- strukturnu vezu ili druge podatke koji ih povezuju sa poznatim neurotoksikantima.
Može da bude i drugih slučajeva u kojima je korišćenje ove metode ispitivanja prikladno (za detalje videti literaturu1).
Ova metoda je razvijena da se prilagodi da zadovolji posebne potrebe za potvrdom specifične histopatološke i bihevioralne neurotoksičnosti hemikalije, kao i da obezbedi karakterizaciju i kvantifikaciju neurotoksičnih reakcija.
U prošlosti se neurotoksičnost izjednačavala sa neuropatijom uključujući neuropatološke lezije ili neurološke disfunkcije kao što su napadi, paraliza ili tremor. Iako je neuropatija važna manifestacija neurotoksičnosti, jasno je da postoje mnogi drugi znaci toksičnosti za nervni sistem (npr. gubitak motorne koordinacije, deficiti čula, disfunkcije učenja i pamćenja) koji se ne moraju oslikavati u neuropatiji ili drugim vrstama studija.
Ova metoda ispitivanja neurotoksičnosti je osmišljena da detektuje glavne neurobihevioralne i neuropatološke efekte kod odraslih glodara. Bihevioralni efekti, čak i odsustvu morfoloških promena, mogu da održavaju štetni uticaj na organizam, ali nisu sve bihevioralne promene specifične za nervni sistem. Zbog toga, bilo koje promene koje su opažene treba oceniti u vezi sa korelativnim histopatološkim, hematološkim ili biohemijskim podacima kao i sa podacima o drugim tipovima sistemske toksičnosti. Ispitivanje koje se obavlja u ovoj metodi je usmereno na davanje karakterizacije i kvantifikacije neurotoksičnih efekata što uključuje specifične histopatološke i bihevioralne postupke koji mogu biti nadalje podržani elektrofiziološkim i/ili biohemijskim istraživanjima1, 2, 3, 4.
Neurotoksikanti mogu da deluju na brojna ciljna mesta u okviru nervnog sistema i putem različitih mehanizama. Nijedan sistem ispitivanja ne može u potpunosti da proceni neurotoksični potencijal svih supstanci, pa može biti korisno iskoristiti druga in vivo ili in vitro ispitivanja koja su specifična za tip opažene ili predviđene neurotoksičnosti.
Ova metoda ispitivanja može da se koristi zajedno sa smernicama datim u OECD vodiču1 kako bi se osmislile studije čija je namera da dalje karakterišu ili povećaju osetljivost kvantifikacije odnosa doza-odgovor kako bi bolje ocenili dozu bez štetnog efekta ili potvrdili poznatu ili verovatnu opasnost od hemikalije. Mogu se osmisliti studije koje identifikuju i ocenjuju neurotoksični mehanizam (mehanizme) ili potkrepljuju već raspoložive podatke korišćenjem postupaka neurobihevioralne i neuropatološke opservacije. Takve studije ne treba da kopiraju podatke koji bi bili generisani korišćenjem standardnih postupaka preporučenih u ovoj metodi. Podaci su već dostupni i nisu neophodni za tumačenje rezultata studije.
Ova metoda ispitivanja neurotoksičnosti, kad se primenjuje sama ili u kombinaciji, daje rezultate koji mogu:
- da identifikuju da li je nervni sistem trajno ili reverzibilno pod uticajem ispitivane hemikalije;
- da doprinesu karakterizaciji promena nervnog sistema koje se povezuju sa izlaganjem hemikaliji i razumevanju relevantnog mehanizma;
- da odrede odnose doza-odgovor i vreme-odgovor kako bi se procenila doza bez štetnog efekta (koja se može koristiti pri uspostavljanju bezbednosnih kriterijuma za hemikaliju).
Ova eksperimentalna metoda koristi oralnu primenu ispitivane supstance. Drugi putevi primene (npr. dermalni ili inhalacioni) mogu biti prikladniji i mogu zahtevati modifikaciju preporučenih postupaka. Razmatranja o izboru puta primene zavise od profila ljudskog izlaganja i raspoloživih toksikoloških ili kinetičkih informacija.
Štetni efekat jeste bilo koja promena u odnosu na osnovno stanje koja je povezana sa tretmanom i koja umanjuje sposobnost organizma da preživi, razmnožava se ili se prilagodi životnoj sredini.
Doza jeste količina primenjene ispitivane supstance. Doza se izražava kao masa (g, mg) ili kao masa ispitivane supstance po jedinici mase eksperimentalne životinje (npr. mg/kg) ili kao konstantna koncentracija u hrani (ppm).
Doziranje jeste opšti pojam koji se sastoji od doze, njene frekvencije i trajanja doziranja.
Neurotoksičnost jeste štetna promena u strukturi ili funkciji nervnog sistema koja je posledica izlaganja hemijskom, biološkom ili fizičkom agensu.
Neurotoksikant jeste bilo koja hemikalija, biološki ili fizički agens koji ima potencijal da uzrokuje neurotoksičnost.
NOAEL jeste skraćenica za dozu bez štetnog efekta (no-observed-adverse effect level) i predstavlja najviši nivo doze pri kome nisu opaženi štetni efekti povezani sa tretmanom.
Ispitivana supstanca se primenjuje oralnim putem u rasponu doza na nekoliko grupa laboratorijskih glodara. Obično su potrebne ponovljene doze i režim doziranja može biti 28 dana, subhronično (90 dana) ili hronično (1 godina ili duže). Postupci opisani u ovoj eksperimentalnoj metodi mogu se koristiti i u studiji akutne neurotoksičnosti. Ispitivanje na životinjama vrši se kako bi se omogućila detekcija ili karakterizacija bihevioralnih i/ili neuroloških abnormalnosti. Raspon ponašanja na koje utiču neurotoksikanti procenjuje se tokom perioda posmatranja. Na kraju ispitivanja, podgrupa životinja oba pola iz svake grupe uzorkuje se in situ i pripremaju se i ispituju preseci mozga, kičmene moždine i perifernih nerava.
Tokom izvođenja studije kao samostalne studije za skrining neurotoksičnosti ili za karakterizaciju neurotoksičnih efekata, životinje u svakoj grupi koje se ne koriste za uzorkovanje i kasnija histopatološka ispitivanja (videti Tabelu 1.) mogu biti korišćene za specifične neurobihevioralne, neuropatološke, neurohemijske ili elektrofiziološke postupke koji mogu zameniti podatke dobijene standardnim ispitivanjima koje zahteva ova metoda1. Ovi dodatni postupci mogu biti posebno korisni kada empirijska posmatranja ili očekivani efekti ukazuju na specifičnu vrstu ili ciljno mesto neurotoksičnosti hemikalije. Preostale životinje se mogu koristiti za procene kao što su one koja zahtevaju metode ispitivanja za studije toksičnosti, ponovljena doza kod glodara.
Kad se postupci iz ove metode ispitivanja kombinuju sa onima iz drugih metoda, potreban je dovoljan broj životinja kako bi se zadovoljili zahtevi za posmatranja iz obe studije.
1.4.1. Izbor životinjske vrste
Preporučena vrsta glodara je pacov, mada se uz opravdanje mogu koristiti i druge vrste. Treba koristiti uobičajene laboratorijske sojeve zdravih mladih odraslih životinje. Koriste se ženke koje nisu ranije rađale i koje nisu trudne. Doziranje uobičajeno treba početi što je ranije moguće posle odbijanja od sise, najbolje ne kasnije nego što životinje navrše šest nedelja i u svakom slučaju pre nego životinje navrše devet nedelja. Kada se ova studija kombinuje sa drugim studijama, ova starost se može prilagoditi. Na početku studije varijacija u telesnoj masi životinja ne sme da prelazi ± 20 % srednje mase za svaki pol. Kada se obavlja kratkoročna studija ponovljene doze kao preliminarna dugoročnoj studiji, u obe studije treba koristiti životinje istog soja i iz istog izvora.
1.4.2. Uslovi smeštaja i ishrane
Temperatura prostorija za eksperimentalne životinje treba da bude 22°C (± 3 °C). Relativna vlažnost treba treba da bude najmanje 30% i da ne prelazi 70% osim za vreme čišćenja prostorija. Ciljna vlažnost treba da bude 50% do 60%. Svetlo treba da bude veštačko sa sekvencama od 12 sati svetla i 12 sati mraka. Buku treba smanjiti na minimum. Može se koristiti konvencionalna laboratorijska ishrana uz neograničene količine vode za piće.
Izbor ishrane može biti uslovljen potrebom da se obezbedi odgovarajuća smeša ispitivane supstance koja se primenjuje ovom metodom. Životinje se smeštaju pojedinačno ili se drže u malim grupama istog pola.
1.4.3. Priprema životinja
Zdrave mlade životinje se slučajnim izborom raspoređuju u grupe. Kaveze treba rasporediti na način da se svedu na minimum mogući efekti zbog njihovog rasporeda. Životinje se jedinstveno identifikuju i drže u kavezima najmanje pet dana pre početka ispitivanja kako bi se aklimatizovale na laboratorijske uslove.
1.4.4. Put primene i priprema doza
Ova metoda ispitivanja posebno se odnosi na oralnu primenu ispitivane supstance. Oralna primena može biti putem prisilnog hranjenja, putem hrane, vode za piće ili kapsulama. Drugi putevi primene (npr. dermalni ili inhalacioni) mogu se koristiti ali zahtevaju modifikaciju preporučenih postupaka. Razmatranja o izboru puta primene zavise od profila izlaganja ljudi i raspoloživih toksikoloških ili kinetičkih podataka. Treba naznačiti objašnjenje za izbor puta primene kao i modifikacije postupaka ove metode ispitivanja.
Kad je neophodno, ispitivana supstanca se može rastvoriti ili suspendovati u odgovarajućem vehikulumu. Preporučuje se korišćenje vodenog rastvora/suspenzije kao prvi izbor, zatim razmatranje primene rastvora/suspenzije u ulju (npr. kukuruzno ulje) i zatim mogući rastvori/suspenzije u drugom vehikulumu. Toksične karakteristike vehikuluma moraju biti poznate. Treba razmotriti i sledeće karakteristike vehikuluma: efekti vehikuluma na apsorpciju, raspodelu, metabolizam ili zadržavanje ispitivane supstance koji mogu izmeniti njene toksične karakteristike; kao i efekte unosa hrane ili vode ili nutritivni status životinja.
1.5.1. Broj i pol životinja
Kada se studija izvodi kao posebna studija, treba koristiti najmanje 20 životinja (10 ženki i 10 mužjaka) u svakoj doznoj i kontrolnoj grupi za ocenjivanja detaljnih kliničkih i funkcionalnih opažanja. Od najmanje pet mužjaka i pet ženki odabranih od tih 10 mužjaka i 10 ženki, treba uzeti uzorke in situ i iste koristiti za detaljnu neurohistopatologiju na kraju studije. U slučajevima kada se na znakove neurotoksičnih efekata posmatra samo ograničen broj životinja u datoj doznoj grupi, treba razmotriti da se te životinje uključe među one odabrane za uzorkovanje. Kada se studija obavlja u kombinaciji sa studijom toksičnosti sa ponovljenom dozom, treba koristiti odgovarajući broj životinja kako bi se zadovoljili ciljevi obe studije. Minimalni broj životinja po grupi za razne kombinacije ispitivanja je prikazan u Tabeli 1. Ukoliko se planiraju žrtvovanja tokom ispitivanja ili ukoliko se planiraju grupe za oporavak za posmatranje reverzibilnosti, perzistencije ili odložene pojave toksičnih efekata posle tretmana ili kada se razmatraju dodatna posmatranja, broj životinja mora biti povećan kako bi se obezbedilo da bude dostupan dovoljan broj životinja potrebnih za posmatranje i histopatologiju.
1.5.2. Tretirana i kontrolna grupa
Treba koristiti najmanje tri dozne i jednu kontrolnu grupu, ali ukoliko se iz procene drugih podataka ne očekuju efekti pri ponovljenoj dozi od 1.000 mg/kg telesna masa/dan, može se obaviti ispitivanje granične doze. Ukoliko nema raspoloživih odgovarajućih podataka, može se izvesti studija traženja raspona kao pomoć pri određivanju doza koje bi se koristile. Osim tretiranja ispitivanom supstancom, sa životinjama u kontrolnoj grupi treba postupati isto kao sa onima iz ispitivane grupe. Ukoliko se pri primeni ispitivane supstance koristi vehikulum, kontrolna grupa treba da primi vehikulum u najvećoj zapremini koja se koristi.
1.5.3. Provera pouzdanosti
Laboratorija koja obavlja studiju treba da navede podatke kojima dokazuje svoju mogućnost da izvede ispitivanje i osetljivost korišćenih postupaka. Takvi podaci treba da pruže dokaze o mogućnosti detekcije i kvantifikacije, kako odgovara, promenama u različitim pokazateljima ispitivanja preporučenim za posmatranje, kao što su autonomni znaci, senzorska reaktivnost, snaga hvatanja i motorna aktivnost. Podaci o hemikalijama koji uzrokuju različite vrste neurotoksičnih efekata i koji se mogu koristiti kao supstance pozitivne kontrole nalaze se u literaturi2, 3, 4, 5, 6, 7, 8, 9. Istorijski podaci mogu se koristiti ukoliko ključni aspekti eksperimentalnih postupaka ostanu isti. Periodično ažuriranje istorijskih podataka se preporučuje. Novi podaci koji pokazuju neprekidnu osetljivost postupaka treba da budu razvijeni kada se promeni neki ključni element izvođenja ispitivanja ili postupka od strane laboratorije.
1.5.4. Izbor doza
Nivoe doza treba izabrati uzimajući u obzir bilo koju ranije uočenu toksičnost i raspoložive kinetičke podatke za ispitivano jedinjenje ili slične materijale. Najvišu dozu treba izabrati sa ciljem izazivanja neurotoksičnih efekata ili jasnih sistemskih toksičnih efekata. Potom treba izabrati opadajući niz doznih nivoa sa ciljem dokazivanja bilo kog odgovora koji je u vezi sa dozom i NOAEL pri najnižem doznom nivou. Doze treba odrediti tako da primarni toksični efekti na nervni sistem mogu da se razlikuju od efekata koji se povezuju sa sistemskom toksičnošću. Dva do tri intervala su često optimum i pored četvrte ispitivane grupe, često se preporučuje da se koriste vrlo veliki intervali (npr. više od faktora 10) između doza. Kada postoji dobra procena ljudske izloženosti, to takođe treba uzeti u obzir.
1.5.5. Ispitivanje granične doze
Ukoliko studija korišćenjem jedne doze od najmanje 1.000 mg/kg TM/dan, korišćenjem opisanih postupaka, ne pokaže nikakve neurotoksične efekte, i ako se toksičnost ne može očekivati na osnovu podataka o strukturno sličnim jedinjenjima, studija u kojoj bi se koristile tri doze ne mora se smatrati neophodnom. Očekivana izloženost ljudi može indikovati potrebu za korišćenjem veće oralne doze u ispitivanju granične doze. Za druge puteve primene kao što su inhalacija ili dermalna primena, fizička i hemijska svojstva ispitivane supstance često mogu diktirati maksimalni nivo izlaganja. Za izvođenje studije akutne oralne toksičnosti, doza za ispitivanje granične doze treba da bude najmanje 2.000 mg/kg.
1.5.6. Primena doza
Životinje se svakodnevno doziraju ispitivanom supstancom, sedam dana nedeljno, tokom perioda od najmanje 28 dana. Korišćenje petodnevnog režima doziranja ili kraćeg izlaganja treba da bude opravdano. Kad se ispitivana supstanca primenjuje prisilnim hranjenjem, isto treba učiniti u jednoj dozi korišćenjem trbušne cevi ili odgovarajućom intubacionom kanulom. Maksimalna zapremina tečnosti koji može biti primenjena zavisi od veličine eksperimentalne životinje. Zapremina ne sme da prelazi 1 ml/100 g TM. Ipak, u slučaju vodenih rastvora može se uzeti u obzir korišćenje do 2 ml/100 g TM. Osim kod iritativnih i korozivnih supstanci koje će uobičajeno pokazati intenzivnije efekte pri višim koncentracijama, varijabilnost u ispitivanoj zapremini treba minimizirati podešavanjem koncentracije kako bi se osigurao konstantna zapremina pri svim nivoima doze.
Za supstance koje se primenjuju putem hrane ili vode za piće, važno je osigurati da količine ispitivane supstance ne ometaju normalnu prehranu ili ravnotežu vode. Kada se ispitivana supstanca primenjuje u hrani bilo kao konstantna prehrambena koncentracija (ppm) ili kada se koristi konstanti dozni nivo u smislu telesne mase životinja, alternativu treba naznačiti. Za supstance koje se primenjuju prisilnim hranjenjem, doza se primenjuje u slično vreme svakog dana i prilagođena je kako bi se zadržao konstantan dozni nivo u smislu telesne mase životinje. Kada se koristi ispitivanje sa ponovljenom dozom kao prethodno ispitivanje za dugoročnu studiju, onda u obe studije treba koristiti sličnu prehranu. Za studije akutne toksičnosti ako nije moguće dati jednu dozu, doza se može davati u manjim delovima tokom perioda koji ne prelaze 24 sata.
1.6.1. Učestalost posmatranja i ispitivanja
Kod studija sa ponovljenom dozom, period posmatranja treba da pokrije period doziranja. Kod studija akutne toksičnosti, treba posmatrati 14-dnevni period posle tretmana. Kod životinja u pratećim grupama koje se drže bez izlaganja za vreme posle tretmana, posmatranja treba da pokriju i ovaj period.
Posmatranja treba obavljati dovoljno često kako bi se povećala verovatnost detekcije bilo kojih bihevioralnih i/ili neuroloških abnormalnosti. Posmatranja treba obavljati po mogućstvu u isto vreme svakog dana uzimajući u obzir period u kome se očekuje pojava efekata posle doziranja. Frekvencija kliničkih posmatranja i funkcionalnih testova je data u Tabeli 2. Ukoliko kinetički ili drugi podaci koji su dobijeni u prethodnim studijama ukazuju na potrebu posmatranja u drugačijim periodima, treba usvojiti alternativni raspored kako bi se prikupio maksimalan broj informacija. Treba dati objašnjenje za promenu rasporeda.
1.6.1.1. Posmatranja opšteg zdravstvenog stanja i mortaliteta/morbiditeta
Zdravstveno stanje svih životinja treba pažljivo posmatrati najmanje jednom dnevno, a dva puta dnevno posmatraju se mortalitet i morbiditet.
1.6.1.2. Detaljna klinička posmatranja
Na životinjama koje su izabrane za tu svrhu obavljaju se detaljna klinička opažanja (videti Tabelu 1.) jednom pre prvog izlaganja (kako bi se omogućila upređivanja kod jedne jedinke) i u različitim intervalima u zavisnosti od trajanja studije (videti Tabelu 2.). Detaljna klinička posmatranja na pratećim grupama treba obaviti na kraju perioda oporavka. Detaljna klinička opažanja se obavljaju izvan kaveza u standardnoj areni. Opažanja se pažljivo beleže korišćenjem sistema ocenjivanja koji uključuje kriterijume ili skale za svaku meru. Laboratorija jasno definiše kriterijume ili skale. Treba uložiti napor kako bi se obezbedilo da varijacije u uslovima ispitivanja budu minimalne (ne sistematično povezane sa tretmanom) i da posmatranja obavljaju obučeni posmatrači koji nisu upoznati sa tretmanom.
Preporučljivo je struktuirano obavljati posmatranja po dobro definisanim kriterijuma (uključujući definiciju "normalnog raspona") i sistematično ih primeniti na svaku životinju u svakom periodu posmatranja. "Normalni raspon" treba adekvatno da se dokumentuje. Beleže se svi uočeni znakovi. Kad god je moguće treba zabeležiti i veličinu uočenih znakova. Klinička opažanja treba da uključuju, ali ne i da budu ograničena na, promene na koži, krznu, očima, sluznicama, pojava sekreta i ekskreta i autonomne aktivnosti (npr. lakrimacija, piloerekcija, veličina zenice, neobičan način disanja i/ili disanje na usta, bilo koje neobične znakove uriniranja ili defekacije i bezbojni urin).
Beleže se i bilo koji neuobičajeni efekti u pogledu položaja tela, nivoa aktivnosti, (npr. povećano ili smanjeno istraživanje standardne arene) i koordinacije pokreta. Promene u držanju (npr. klaćenje, ataksija), stavu (npr. pogrbljenost) i reakciji na postupanje, smeštanje ili drugi stimulusi okoline kao i prisustvo kloničnih ili toničnih pokreta, konvulzija ili tremora, stereotipa (npr. preterano timarenje, neobični pokreti glave, ponavljajuće kretnje u krug) ili čudno ponašanje (npr. griženje ili preterano lizanje, samosakaćenje, hod unazad, vokalizacija) ili agresija takođe se beleže.
1.6.1.3. Funkcionalna ispitivanja
Slično detaljnim kliničkim posmatranjima, funkcionalna ispitivanja takođe treba obavljati pre izlaganja, kao i često posle toga, na svim životinjama koje su izabrane za tu svrhu (videti Tabelu 1.). Učestalost funkcionalnih ispitivanja takođe zavisi od trajanja studije (videti Tabelu 2.). Pored perioda posmatranja iz Tabele 2, treba obavljati funkcionalna ispitivanja na pratećim grupama sve do usmrćenja. Funkcionalna ispitivanja treba da uključuju senzornu reaktivnost na stimuluse raznih vrsta (npr. zvučni, vizuelni i proprioceptivni stimulusi5, 6, 7 ocenjivanje snage stiska8 i ocenjivanje motorne aktivnosti9). Motornu aktivnost treba meriti automatskom spravom kojom je moguće detektovati smanjenu i povećanu aktivnost. Ukoliko se koristi drugi definisani sistem, on mora biti kvantitativan i njegova osetljivost i pouzdanost treba pokazati. Svaki uređaj treba testirati kako bi se osigurala pouzdanost tokom vremena i konzistentnost između uređaja. Više detalja o postupcima koji se mogu pratiti nalazi se u literaturi. Ukoliko nema podataka (npr. struktura - aktivnost, epidemiološki podaci, druge studije toksičnosti) koji bi ukazivali na potencijalne neurotoksične efekte, razmatra se uključivanje specijalizovanijih testova senzorne ili motorne funkcije ili učenja i pamćenja kako bi se detaljnije proučili ti mogući efekti. Više podataka o specijalizovanim testovima dato je u literaturi1.
Izuzetno, životinje koje pokazuju znakove toksičnosti u obimu koji značajno ometa funkcionalno ispitivanje mogu biti izostavljene iz tog ispitivanja. Treba dati opravdanje za isključivanje tih životinja iz funkcionalnog ispitivanja.
1.6.2. Telesna masa i potrošnja hrane/vode
Za studije koje traju do 90 dana sve životinje treba meriti najmanje jednom nedeljno i treba izmeriti potrošnju hrane (potrošnju vode kada se ispitivana supstanca unosi tim putem) takođe najmanje jednom nedeljno. Za dugoročne studije sve životinje treba meriti najmanje jednom nedeljno prvih 13 nedelja, a potom najmanje jednom na svake 4 nedelje. Treba meriti potrošnju hrane (potrošnju vode kada se ispitivana supstanca unosi tim putem) takođe najmanje jednom nedeljno tokom prvih 13 nedelja, a kasnije u približno tromesečnim intervalima osim ukoliko zdravstveno stanje ili promena telesne mase ne nalažu drugačije.
1.6.3. Oftalmološka ispitivanja
Za studije koje traju duže od 28 dana treba obaviti oftalmološki pregled uz korišćenje oftalmoskopa ili sličnog odgovarajućeg instrumenta, pre primene ispitivane supstance i po završetku studije, najbolje na svim životinjama, ali najmanje na onima iz grupa visoke doze i iz kontrolne grupe.
Ukoliko se primete promene u očima ili ukoliko klinički znaci ukazuju na potrebu, treba pregledati sve životinje. Kod dugoročnih studija oftalmološki pregled treba obavljati u 13. nedelji. Oftalmološki pregledi se ne moraju obavljati ukoliko su ovi podaci već dostupni iz drugih studija sličnog trajanja i sličnih doznih nivoa.
1.6.4. Hematološka i medicinsko-biohemijska ispitivanja
Kada se izvodi studija neurotoksičnosti u kombinaciji sa studijom sistemske toksičnosti sa ponovljenom dozom, treba uraditi hematološka ispitivanja i medicinsko-biohemijska određivanja kako je izloženo u metodi ispitivanja sistemske toksičnosti. Prikupljanje uzoraka treba obaviti tako da bilo koji potencijalni efekti na neuropatologiju budu svedeni na minimum.
1.6.5. Histopatološka ispitivanja
Neuropatološki pregled treba da bude osmišljen tako da dopuni i proširi opažanja iz in vivo faze studije. Tkiva najmanje 5 životinja/pol/grupa (videti Tabelu 1. i sledeći pasus ovog odeljka) treba fiksirati in situ, korišćenjem opštepriznatih tehnika perfuzije i fiksacije (videti literaturu3, i literaturu4). Bilo koje uočljive promene treba zabeležiti. Kad se studija izvodi kao samostalna studija za proveru neurotoksičnosti ili za karakterizaciju neurotoksičnih efekata, preostale životinje mogu biti korišćene bilo za specifične neurobihevioralne10, 11, neuropatološke10, 11, 12, 13, neurohemijske10, 11, 14, 15 ili elektrofiziološke10, 11, 16, 17 postupke kao dopuna postupcima i ispitivanjima koji su ovde opisani ili za povećanje broja jedinki pregledanih za histopatologiju. Ovi dodatni postupci su od posebne koristi kad empirijska opažanja ili očekivani efekti ukazuju na specifični tip ili ciljno mesto neurotoksičnosti2, 3. Alternativno, preostale životinje mogu da se podvrgnu rutinskom patološkom ocenjivanju kako je opisano u metodi za studije sa ponovljenom dozom.
Opšti postupak bojenja kao što je hematoksilin i eozin (u daljem tekstu: H&E) treba obaviti za sve uzroke tkiva u parafinu i treba izvesti mikroskopski pregled. Ukoliko se uoče znaci periferne neuropatije ili ako se na iste sumnja, potrebno je ispitati uzorke perifernog nervnog tkiva u plastici. Klinički znaci takođe mogu naznačiti dodatna mesta za ispitivanje ili korišćenje posebnih postupaka bojenja. Uputstvo o dodatnim mestima dato je u literaturi3, 4. Odgovarajuće specijalne boje za pokazivanje specifičnih tipova patoloških promena mogu biti od pomoći18.
Na reprezentativnim presecima centralnog i perifernog nervnog sistema treba izvršiti histološki pregled (videti u literaturi3 i literaturi4). Ispitivana područja obično uključuju: prednji mozak, središte cerebruma, uključujući presek kroz hipokampus, srednji mozak, cerebelum, pons, produžena moždina, oko sa optičkim nervom i retinom, kičmenu moždinu i cervikalna i lumbalna zadebljanja, dorzalni koren ganglia, dorzalna i ventralna vlakna korenova, proksimalni išijasni nerv, proksimalni tibijalni nerv (u kolenu) i grane tibijalnog nerva za mišić lista. Preseci kičmene moždine i perifernih nerava treba da uključe i poprečne ili transverzalne i uzdužne preseke. Treba obratiti pažnju na vaskulaturu nervnog sistema. Uzorak skeletnog mišića, posebno mišića lista, takođe treba pregledati. Posebnu pažnju treba obratiti na mesta sa celularnom ili fibroznom strukturom i uzorak CNS i PNS za koji se zna da je posebno pogođen neurotoksikantima.
Smernice o neuropatološkim promenama koje tipično proizilaze iz izlaganja toksikantu date su u literaturi3, 4. Preporučuje se postepeno ispitivanje uzoraka tkiva u kome se preseci grupe visoke doze prve upoređuju sa onima iz kontrolne grupe. Ukoliko se ne uoče neuropatološke promene u uzorcima iz ovih grupa, dalja analiza nije potrebna. Ukoliko se uoče neuropatološke promene u grupi visoke doze, uzorak svakog potencijalno pogođenog tkiva iz srednjih i nižih doznih grupa treba kodirati i redom ispitati.
Ukoliko se pronađu bilo kakve neuropatološke promene u kvalitativnoj analizi, treba obaviti drugo ispitivanje na svim područjima nervnog sistema koji pokazuju takve promene. Preseke iz svih doznih grupa svih potencijalno zahvaćenih regija treba kodirati i ispitati nasumce bez poznavanja koda. Frekvenca i ozbiljnost svake lezije treba da budu zabeležene. Pošto se ocene sve regije iz svih grupa kod se može dešifrovati i može se obaviti statistička analiza kako bi se ocenio odnos doza-odgovor. Primere različitih stepena ozbiljnosti svake lezije treba opisati.
Neuropatološki nalazi treba da se ocene u kontekstu bihejvioralnih posmatranja i merenja, kao i drugih podataka iz prethodnih i istovremenih studija sistemske toksičnosti ispitivane supstance.
Navode se pojedinačni podaci. Svi podaci se navode tabelarno. Za svaku ispitivanu ili kontrolnu grupu navode se: broj životinja na početku ispitivanja, broj životinja koje su pronađene mrtve za vreme ispitivanja ili usmrćene iz humanih razloga, kao i vreme smrti ili humanog žrtvovanja, broj koji pokazuje znakove toksičnosti, opis znakova toksičnosti koji su uočeni, uključujući vreme nastanka, trajanje, vrstu i ozbiljnost bilo kojih toksičnih efekata, broj životinja koje pokazuju lezije, uključujući vrstu i ozbiljnost lezije (lezija).
2.2. PROCENA I TUMAČENJE REZULTATA
Nalazi studije se procenjuju u pogledu incidence, ozbiljnosti i korelacije neurobihevioralnih i neuropatoloških efekata (neurohemijskih ili elektrofizioloških efekata takođe, ukoliko su uključena dodatna ispitivanja) ili bilo kojih drugih uočenih štetnih efekata. Kada je moguće, numerički rezultati se procenjuju odgovarajućom pšteprihvaćenom statističkom metodom. Statističke metode se biraju prilikom osmišljavanja studije.
Izveštaj o ispitivanju mora da sadrži podatke o:
1) Ispitivanoj supstanci:
- fizička priroda (uključujući izomeriju, čistoću i fizička i hemijska svojstva);
- identifikacioni podaci.
2) Vehikulumu (ako odgovara):
- opravdanje za izbor vehikuluma.
3) Eksperimentalnim životinjama:
- korišćena vrsta/soj;
- broj, starost i pol životinja;
- izvor, uslovi smeštaja, aklimatizacija, ishrana, itd.;
- pojedinačne mase životinja na početku ispitivanja.
4) Uslovima ispitivanja:
- detalji o formulaciji ispitivane supstance/priprema ishrane, postignuta koncentracija, stabilnost i homogenost preparata:
- specifikacija primenjenih doza, uključujući detalje o vehikulumu, zapremini i fizičkom obliku primenjenog materijala;
- detalji o primeni ispitivane supstance;
- obrazloženje za izabrane nivoe doza;
- obrazloženje za put primene i trajanje izlaganja;
- konverzija koncentracije ispitivane supstance (ppm) iz ishrane/vode za piće u stvarnu dozu (mg/kg telesna masa / dan), ukoliko je primenjivo;
- detalji o kvalitetu hrane i vode.
5) Postupcima ispitivanja i posmatranja:
- detalji o svrstavanju životinja svake grupe u podgrupe za perfuziju;
- detalji o sistemu bodovanja, uključujući kriterijume i skale za svako merenje u detaljnim kliničkim posmatranjima;
- detalji o funkcionalnim ispitivanjima za senzornu reaktivnost na stimuluse različitih vrsta (npr. zvučni, vizuelni i proprioceptivni), za ocenjivanje snage stiska, za ocenjivanje motorne aktivnosti (uključujući detalje o automatskim spravama za detekciju aktivnosti), i drugim korišćenim postupcima;
- detalji o oftalmološkim pregledima i ako odgovara hematološkim ispitivanjima i medicinsko-biohemijskim ispitivanjima sa relevantnim osnovnim vrednostima;
- detalji specifičnih neurobihevioralnih, neuropatoloških, neurohemijskih ili elektrofizioloških postupaka.
6) Rezultatima:
- telesna masa/promene telesne mase, uključujući telesnu masu pri žrtvovanju;
- potrošnja hrane i vode, ukoliko je primenljivo;
- podaci o toksičnim efektima po polu i doznom nivou, uključujući znake toksičnosti ili smrtnost;
- priroda, ozbiljnost i trajanje (vreme nastanka i kasniji tok) detaljnih kliničkih nalaza (prolaznih ili ne);
- detaljan opis svih rezultata funkcionalnih ispitivanja;
- nalazi obdukcije;
- detaljan opis svih specifičnih neurobihevioralnih, neuropatoloških, neurohemijskih ili elektrofizioloških nalaza, ukoliko je primenljivo;
- podaci o apsorpciji i metabolizmu, ukoliko su dostupni;
- statistička obrada rezultata, kad je primenjivo.
7) Obrazloženju rezultata:
- podatke o efektu doze;
- odnos drugih toksičnih efekata do zaključka o neurotoksičnom potencijalu ispitivane supstance;
- doza bez štetnog efekta (NOAEL).
8) Zaključku:
- preporučuje specifičan izveštaj o celokupnoj neurotoksičnosti ispitivane hemikalije.
1. OECD Giudance Document on Neurotoxicity Testing Strategies and Test Methods. OECD, Paris, InPreparation.
2. Test Guideline for a Developmental Neurotoxicity Study, OECD Guidelines for the Testing of Chemicals. In preparation.
3. World Health Organisation (WHO) (1986) Environmental Health Criteria document 60: Principles and Methods for the Assessment of Neurotoxicity associated with Exposure to Chemicals.
4. Spencer, P.S. and Schaumburg, H.H. (1980) Experimental and Clinical Neurotoxicology. Eds. Spencer, P.S. and Schaumburg, H.H. eds. Williams and Wilkins, Baltimore/London.
5. Tupper, D.E. and Wallace, R.B., (1980) Utility of the Neurological Examination in Rats. Acta Neurobiol. Exp., 40, p. 999-1003.
6. Gad, S.C., (1982) A Neuromuscular Screen for Use in Industrial Toxicology. J. Toxicol. Environ. Health, 9, p. 691-704.
7. Moser, V.C., McDaniel, K.M. and Phillips, P.M., (1991) Rat Strain and Stock Comparisons Using a Functional Observational Battery: Baseline Values and Effects of amitraz. Toxic. Appl. Pharmacol., 108, p. 267-283.
8. Meyer, O.A., Tilson, H.A., Byrd, W.C. and Riley, M.T., (1979) A Method for the Routine Assessment of Fore- and Hind- limb Grip Strength of Rats and Mice. Neurobehav. Toxicol., 1, p. 233-236.
9. Crofton, K.M., Haward, J.L., Moser, V.C., Gill, M.W., Reirer, L.W., Tilson, H.A. and MacPhail, R.C. (1991) Interlaboratory Comparison of Motor Activity Experiments: Implication for Neurotoxicological Assessments. Neurotoxicol. Teratol., 13, p. 599-609.
10. Tilson, H.A., and Mitchell, C.L. eds., (1992) Neurotoxicology Target Organ Toxicology Series. Raven Press, New York.
11. Chang, L.W., ed., (1995) Principles of Neurotoxicology. Marcel Dekker, New York.
12. Broxup, B., (1991) Neuopathology as a screen for Neurotoxicity Assessment. J. Amer. Coll. Toxicol., 10, p. 689-695.
13. Moser, V.C., Anthony, D.C., Sette, W.F. and MacPhail, R.C., (1992) Comparison of Subchronic Neurotoxicity of 2-Hydroxyethyl Acrylate and Acrylamide in Rats. Fund. Appl.Toxicol., 18, p. 343-352.
14. O‘Callaghan, J.P. (1988). Neurotypic and Gliotypic Proteins as Biochemical Markers of Neurotoxicity. Eurotoxicol. Teratol., 10, p. 445-452.
15. O‘Callaghan J.P. and Miller, D.B., (1988) Acute Exposure of the Neonatal Rat to Triethyltin Results in Persistent Changes in Neurotypic and Gliotypic Proteins. J. Pharmacol. Exp. Ther., 244, p. 368-378. L 142/428 EN Official Journal of the European Union 31.5.2008.
16. Fox. D.A., Lowndes, H.E. and Birkamper, G.G., (1982) Electrophysiological Techniques in Neurotoxicology. In: Nervous System Toxicology. Mitchell, C.L. ed. Raven Press, New York, p. 299-335.
17. Johnson, B.L., (1980) Electrophysiological Methods in neurotoxicity Testing. In: Experimental and Clinical Neurotoxicology. Spencer, P.S. and Schaumburg, H.H. eds., Williams and Wilkins Co., Baltimore/ London, p. 726-742.
18. Bancroft, J.D. and Steven A., (1990) Theory and Pratice of Histological Techniques. Chapter 17, Neuropathological Techniques. Lowe, James and Cox, Gordon eds. Churchill Livingstone.
Tabela 1.: Minimalni broj životinja po grupi kada se studija neurotoksičnosti izvodi odvojeno ili u kombinaciji s drugim studijama
| STUDIJA NEUROTOKSIČNOSTI SPROVEDENA KAO | |||
Posebna studija | Kombinovana sa | Kombinovana sa | Kombinovana sa studijom hronične toksičnosti | |
Ukupan broj životinja po grupi | 10 mužjaka i | 10 mužjaka i | 15 mužjaka i | 25 mužjaka i |
Broj životinja odabranih za funkcionalna ispitivanja uključujući detaljno kliničko posmatranje | 10 mužjaka i | 10 mužjaka i | 10 mužjaka i | 10 mužjaka i |
Broj životinja odabran za uzorkovanje in situ i neurohistopatologiju | 5 mužjaka i | 5 mužjaka i | 5 mužjaka i | 5 mužjaka i |
Broj životinja odabran za posmatranja toksičnosti ponovljene doze/subhronične/hronične, hematologiju, medicinsku biohemiju, histopatologiju, itd. kako je naznačeno u Vodiču |
| 5 mužjaka i 5 ženkiLIII | 10 mužjakaXVI | 20 mužjakaXVI |
Dodatna posmatranja, kada je odgovarajuće | 5 mužjaka i |
|
|
|
____________
LIII Uključuje pet životinja odabranih za funkcionalna ispitivanja i detaljno kliničko posmatranje kao deo studije neurotoksičnosti.
Tabela 2.: Frekvenca kliničkog posmatranja i funkcionalnih ispitivanja
Vrsta posmatranja | Trajanje studije | ||||
Akutno | 28-dana | 90-dana | Hronično | ||
Kod svih životinja | Opšte zdravstveno stanje | dnevno | dnevno | dnevno | dnevno |
Mortalitet /morbiditet | Dva puta dnevno | Dva puta dnevno | Dva puta dnevno | Dva puta dnevno | |
Kod životinja odabranih za funkcionalna posmatranja | Detaljna klinička posmatranja | - pre prvog izlaganja | - pre prvog izlaganja | - pre prvog izlaganja | - pre prvog izlaganja |
Funkcionalna ispitivanja | - pre prvog izlaganja | - pre prvog izlaganja | - pre prvog izlaganja | - pre prvog izlaganja | |
B.44. APSORPCIJA PREKO KOŽE: IN VIVO METODA
Ova metoda potpuno odgovara metodi OECD: TG 427 (2004).
Ekspozicija mnogim hemikalijama se dešava uglavnom putem kože dok se većina studija izvodi na laboratorijskim životinjama peroralnom primenom. In vivo ispitivanje perkutane apsorpcije navedeno u ovoj smernici obezbeđuje vezu koja je neophodna za ekstrapolaciju iz studija oralne toksičnosti kada se radi procena bezbednosti posle dermalne ekspozicije.
Supstanca mora da prođe veliki broj slojeva ćelija kože pre nego što dospe u cirkulaciju. Za većinu supstanci sloj koji ograničava brzinu prolaza kroz kožu je stratum corneum koji se sastoji od mrtvih ćelija. Permeabilnost kroz kožu zavisi kako od lipofilnosti hemikalije tako i od debljine spoljašnjeg sloja epidermisa, kao i od faktora kao što su molekulska masa i koncentracija supstance. Uglavnom je koža pacova i zečeva permeabilnija od ljudske, dok je permeabilonost kože zamorčića i majmuna sličnija ljudskoj.
Metode za merenje perkutane apsorpcije mogu da se podele u dve kategorije: in vivo i in vitro. In vivo metoda može da obezbedi dobre podatke o apsorpciji kroz kožu kod različitih vrsta.
Nedavno su razvijene in vitro metode. One koriste transport kroz ceo ili delimični sloj životinjske ili ljudske kože u rezervoar sa tečnošću. In vitro metoda se opisuje u posebnoj metodi ispitivanja1. Preporučuje se da se koristi OECD vodič2 kao pomoć u izboru odgovarajuće metode ispitivanja apsorpcije kroz kožu za datu situaciju s obzirom da se u ovom vodiču nalaze detalji o pogodnosti kako in vitro tako i in vivo metoda.
In vivo metoda opisana u ovoj metodi omogućuje određivanje penetracije ispitivane supstance kroz kožu u sistemski odeljak. Ova tehnika se široko koristi tokom više godina3, 4, 5, 6, 7. Iako u mnogim slučajevima in vitro studije perkutane apsorpcije mogu da budu pogodne, postoje mnoge situacije u kojima samo in vivo studije mogu da obezbede neophodne podatke.
Prednost in vivo metode je da koristi fiziološki i metabolički intaktan sistem, koristi vrste uobičajene za mnoge studije toksičnosti i može da se modifikuje za primenu na drugim vrstama. Nedostaci su korišćenje živih životinja, potreba za korišćenjem radioaktivno obeleženog materijala da bi se dobili pouzdani rezultati, teškoće u određivanju rane faze apsorpcije i razlike u permeabilnosti kože vrsta koja se najčešće koristi (pacova) i kože čoveka.
Životinjska koža je uglavnom permeabilnija i zbog toga na osnovu ovih ispitivanje može da se preceni apsorpcija kroz kožu čoveka6, 8, 9. Kausitične/korozivne supstance ne treba ispitivati na živim životinjama.
Neapsorbovana doza predstavlja dozu koja je isprana sa površine kože posle ekspozicije i deo koji je prisutan na neokluzivnom pokrivaču, uključujući i dozu za koju je pokazano da je isparila sa kože tokom ispitivanja.
Apsorbovana doza (in vivo) obuhvata dozu koja se nalazi u urinu, u ispirku kaveza, fecesu, izdahnutom vazduhu (ako je mereno), krvi, tkivima (ako su sakupljana) i u lešu životinje posle uklanjanja dela kože na koju je primenjena supstanca.
Apsorbabilna doza predstavlja dozu u ili na koži posle ispiranja.
Ispitivana supstanca (poželjno je da bude radioaktivno obeležena) primenjuje se na ošišanu kožu životinja u jednom ili više doznih nivoa u obliku reprezentativnih preparata u upotrebi. Ispitivani preparati se mogu ostaviti u kontaktu sa kožom određeno vreme pod odgovarajućim pokrivačem (ne-okluzivni, semi-okluzivni ili okluzivni) da bi se sprečila ingestija ispitivanog preparata. Na kraju ekspozicije pokrivač se uklanja i koža čisti odgovarajućim sredstvom za čišćenje, pokrivač i materijal za čišćenje se čuvaju za analizu, a na kožu se nanosi novi pokrivač. Životinje se smeštaju u pojedinačne metaboličke kaveze i ekskreti i izdahnuti vazduh se skupljaju za analizu, pre, tokom i posle perioda ekspozicije. Sakupljanje izdahnutog vazduha može da se izostavi ako postoji dovoljno podataka da je formiranje ispraljivih radioaktivnih metabolita minimalno ili se uopšte ne javlja. Svaka studija obično uključuje nekoliko grupa životinja koje će biti izložene ispitivanom preparatu. Jedna grupa će biti žrtvovana na kraju perioda ekspozicije. Potom će druge grupe biti žrtvovane u planiranim intervalima2. Na kraju perioda sakupljanja uzoraka, ostatak životinja se žrtvuje, sakuplja se krv za analizu, uklanja se i čuva za analizu deo kože na koju je ispitivana supstanca primenjena, a leševi se analiziraju za svaki neizlučeni materijal. Uzorci se analiziraju na odgovarajuće načine i određuje se stepen perkutane apsorpcije6, 8, 9.
1.4.1. Izbor životinjske vrste
Obično se koristi pacov, ali sojevi bez dlake i vrste koje imaju brzine apsorpcije sličnije onoj kod ljudi mogu takođe da se koriste3, 6, 7, 8, 9. Uključuju se mlade, odrasle i zdrave životinje istog pola (obično muškog pola) uobičajenih laboratorijskih sojeva. Na početku studije odstupanje u telesnoj masi životinje ne treba da bude veće od ± 20% od prosečne mase. Na primer, mužjaci pacova od 200 g do 250 g, posebno na gornjoj polovini ovog raspona.
1.4.2. Broj i pol životinja
Grupa od najmanje četiri životinje istog pola treba da se koristi za svaki ispitivani preparat i svako planirano vreme žrtvovanja. Svaka grupa životinja će biti žrtvovana posle različitih vremenskih intervala, npr. na kraju perioda ekspozicije (obično 6 ili 24 sata) i u narednim prilikama (npr. 48 i 72 sata). Ako postoje podaci koji pokazuju suštinske razlike u dermalnoj toksičnosti između mužjaka i ženki, treba koristiti osetljiviji pol. Ako ne postoje takvi podaci, može da se koristi bilo koji od polova.
1.4.3. Uslovi smeštaja i ishrane
Temperatura u prostoru za čuvanje eksperimentalnih životinja treba da bude 22 °C (± 3 °C). Relativna vlažnost treba da bude najmanje 30% i da ne premašuje 70% osim za vreme čišćenja prostora. Ciljna vlažnost treba da bude 50% do 60%. Osvetljenje treba da bude veštačko sa periodom svetlosti od 12 sati i periodom mraka od 12 sati.
Za ishranu može da se koristi uobičajena hrana za eksperimentalne životinje, koja treba da bude slobodno dostupna životinjama uz neograničen pristup vodi za piće. Za vreme studije, a po mogućstvu i za vreme aklimatizacije, životinje se smeštaju u pojedinačne metaboličke kaveze. S obzirom da prosipanje hrane i vode mogu da kompromituju rezultate, verovatnoća za ovakve događaje treba da se minimalizuje.
1.4.4. Priprema životinja
Životinje se označavaju da bi mogle da se pojedinačno identifikuju i čuvaju se u kavezima najmanje pet dana pre početka studije da bi se aklimatizovale na laboratorijske uslove.
Posle aklimatizacije i približno 24 sata pre doziranja, region ramena i leđa svake životinje se ošiša. Propustljivost oštećene kože je različita od one kod intaktne kože i zbog toga se izbegava abrazija kože. Posle šišanja i približno 24 sata pre primene ispitivane supstance na kožu (videti odeljak 1.4.7. ove metode), površina kože treba da se obriše acetonom da bi se uklonio sebum. Dodatno pranje sapunima i vodom se ne preporučuje zbog toga što rezidue sapuna mogu da promovišu apsorpciju ispitivane supstance. Površina kože mora da bude dovoljno velika da obezbedi pouzdano izračunavanje apsorbovane količine ispitivane hemikalije po cm2 kože, poželjno je najmanje 10 cm2. Ova površina je praktična za pacove od 200 g do 250 g TM. Posle pripreme životinje se vraćaju u metaboličke kaveze.
1.4.5. Ispitivana supstanca
Ispitivana supstanca je entitet čija penetracione karakteristike treba da se ispitaju. Idealno, ispitivana supstanca treba da bude radioaktivno obeležena.
1.4.6. Ispitivani preparat
Preparat ispitivane supstance (npr. čista, razblažena hemikalija ili formulisani materijal koji sadrži ispitivanu hemikaliju koja se primenjuje na kožu) treba da bude isti (ili realni surogat) kao onaj kojem će biti izloženi ljudi ili druga ciljna vrsta. Svako odstupanje od preparata u upotrebi mora biti opravdano. Kada je neophodno, ispitivna supstanca se rastvara ili suspenduje u odgovarajućem vehikulumu. Za vehikulume koji nisu voda treba poznavati apsorpcione karakteristike i moguće interakcije sa ispitivanom supstancom.
1.4.7. Primena na kožu
Na koži se definiše određeni deo površine za aplikaciju. Poznata količina ispitivanog preparata se ravnomerno nanosi na to mesto. Ova količina obično treba da imitira moguću ekpoziciju kod ljudi, tipično 1 mg/cm2 do 5 mg/cm2 za čvrste ili do 10 µl/cm2 za tečnosti. Sve druge količine treba da se opravdaju uslovima očekivane primene, ciljevima studije ili fizičkim svojstvima ispitivanog preparata. Posle aplikacije, tretirano mesto mora da bude zaštićeno od lizanja. Primer tipične sprave dat je na Slici 1. Obično se mesto primene zaštiti neokluzivnim pokrivačem (npr. pokrivač od permeabilne najlonske gaze). Za dužu primenu mesto primene treba da bude pokriveno. U slučaju isparavanja poluisparljivih ispitivanih supstanci, dolazi do neprihvatljivog smanjenja recovery vrednosti za ispitivanu supstancu (videti odeljak 1.4.10. prvi pasus ove metode), neophodno je da se isparena supstanca uhvati filterom od medicinskog uglja koji pokriva spravu za primenu (videti Sliku 1). Važno je da ni jedna sprava ne oštećuje kožu, niti da apsorbuje, niti da reaguje sa ispitivanim preparatom. Životinje se vraćaju u pojedinačne metaboličke kaveze da bi se sakupljali ekskreti.
1.4.8. Trajanje ekspozicije i uzorkovanje
Trajanje ekspozicije je vremenski interval između primene i uklanjanja pranjem ispitivanog preparata sa kože. Treba da se koristi odgovarajući period ekspozicije (obično 6 ili 24 sata), što se zasniva na očekivanoj dužini ekspozicije kod ljudi. Posle perioda ekspozicije, životinje se zadržavaju u metaboličkim kavezima sve do unapred planiranog žrtvovanja. Životinje se posmatraju u redovnim intervalima tokom studije da bi se utvrdili znaci toksičnih/abnormalnih odgovora. Na kraju perioda ekspozicije, tretiranu kožu treba pregledati da bi se proverilo da li postoje vidljivi znaci iritacije.
Metabolički kavezi treba da omoguće odvojeno sakupljanje urina i fecesa tokom studije. Oni treba da obezbede i sakupljanje 14C -ugljendioksida i isparljivih 14C- ugljenikovih jedinjenja koja treba da se analiziraju kada su proizvedena u dovoljnoj količini (više od 5%). Urin, feces i tečnost za hvatanje (npr. 14C- ugljendioksida i isparljivih 14C- ugljenikovih jedinjenja) treba pojedinačno sakupljati od svake grupe u svako vreme uzimanja uzoraka. Ako postoji dovoljno podataka da se formira malo ili nimalo isparljivog, radioaktivnog metabolita, mogu da se koriste otvoreni kavezi.
Ekskreti se sakupljaju za vreme perioda ekspozicije i do 24 sata posle inicijalnog kontakta sa kožom, a potom svakodnevno do kraja ispitivanja. Mada su tri intervala sakupljanja ekskreta obično dovoljna, predviđena namena ispitivanog preparata ili postojeći kinetički podaci mogu da ukažu na to da je za studiju pogodnije da se uključi dodatno vreme i intervali.
Na kraju perioda ekspozicije, zaštitna naprava se uklanja sa svake životinje i odvojeno zadržava za analizu. Tretirana koža svih životinja treba da se opere najmanje tri puta odgovarajućim sredstvom za čišćenje koristeći pogodne ubruse. To mora da se čini pažljivo da bi se izbegla kontaminacija drugih delova tela životinje. Sredstvo za čišćenje mora da bude deo uobičajene higijenske prakse, npr. vodeni rastvor sapuna. Na kraju koža treba da se osuši. Svi ubrusi i ispirci moraju da se sačuvaju za analizu. Pre vraćanja u kaveze, treba da se primeni nov pokrivač da bi se zaštitilo tretirano mesto kod životinja koje su predviđene za kasnije ispitivanje.
1.4.9. Postupci žrtvovanja
Za svaku grupu, pojedinačne životinje se žrtvuju u predviđeno vreme i krv se sakuplja za analizu. Zaštitna naprava ili pokrivač treba da se uklone za analizu. Koža sa mesta aplikacije i sličnog dela nedozirane, ošišane kože treba da se ukloni od svake životinje za odvojenu analizu. Mesto primene može da se razdeli da bi se odvojio stratum corneum od epidermisa kako bi se dobio podatak o dispoziciji ispitivane hemikalije. Određivanje ove raspodele tokom vremena, posle perioda ekspozicije treba da ukaže na sudbinu ispitivane hemikalije u stratum cornerumu. Da bi se olakšalo razdeljivanje kože (posle završenog pranja kože i žrtvovanja životinje), svi zaštitni pokrivači se uklanjaju. Koža mesta aplikacije sa prstenom okolne kože se odvaja od pacova i pričvršćuje iglama za podlogu. Parče adhezivne trake se primenjuje na površinu kože uz blagi pritisak, a potom se traka uklanja zajedno sa delom stratum corneuma. Zatim se primenjuju novi komadi adhezivne trake sve dok se traka više ne lepi za površinu kože, kada je sav stratum corneum uklonjen. Za svaku životinju svi delovi adhezivne trake mogu da se čuvaju u jednom kontejneru kome je dodat tkivni digestant da bi se solubilizovao stratum corneum. Sva potencijalno ciljna tkiva mogu da se uklone za posebna merenja pre nego što se ostaci analiziraju za apsorbovanu lešnu dozu. Leševi pojedinačnih životinja treba da se sačuvaju za analizu. Obično je dovoljna analiza ukupnog sadržaja. Ciljni organi mogu da se uklone za odvojene analize (ako su na to ukazale druge studije). Urin koji se nalazi u mokraćnoj bešici pri žrtvovanju, treba da se doda u prethodno sakupljen urin. Posle skupljanja ekskreta iz metaboličkih kaveza u vreme žrtvovanja životinja, kavezi i uređaji za sakupljanje treba da se operu odgovarajućim rastvaračem. Drugi delovi opreme koji mogu biti kontaminirani takođe treba da se analiziraju.
1.4.10. Analize
U svim studijama treba postići odgovarajuću recovery vrednost (npr. srednju vrednost od 100% ± 10% radioaktivnosti). Recovery vrednosti van ovih moraju se opravdati. Količina primenjene doze u svakom uzorku treba da se analizira odgovarajućim validovanim postupcima.
Statistička razmatranja treba da uključe meru varijacije za ponavljanja za svaku primenu.
Sledeća merenja treba da se izvrše za svaku životinju, za svako vreme uzorkovanja, za ispitanu hemikaliju i/ili metabolite (uz pojedinačne podatke, podaci grupisani prema vremenu uzorkovanja treba da se prikažu kao srednje vrednosti):
- količina udružena sa zaštitnim napravama;
- količina koja može da se ukloni sa kože;
- količina u/na koži koja ne može da se ispere sa kože;
- količina u uzorcima krvi;
- količina u ekskretima i izdahnutom vazduhu (ako je primenljivo);
- količina koja ostane u lešu ili u organima koji su uklonjeni za odvojenu analizu.
Količina ispitivane supstance i/ili metabolita u ekskretima, izdahnutom vazduhu, krvi i lešu omogućava određivanje ukupne apsorbovane količine u svakoj vremenskoj instanci. Može se izračunati i količina ispitivane hemikalije koja je apsorbovana po cm2 izložene kože za vreme perioda ekspozicije.
Izveštaj o ispitivanju mora da sadrži zahteve predviđene u protokolu, uključujući opravdanost sistema za ispitivanje koji je korišćen, a mora da sadrži i podatke o:
1) Ispitivanoj supstanci:
- podaci za identifikaciju (npr. CAS broj ako je dostupan, izvor, čistoća (radiohemijska čistoća), poznate nečistoće, broj serije);
- fizička svojstva, fizička i hemijska svojstva (npr. pH, isparljivost, rastvorljivost, stabilnost, molekulska masa i log Pow).
2) Ispitivanom preparatu:
- formulacija i opravdanost primene;
- detalji o ispitivanom preparatu, primenjena količina, postignuta koncentracija, vehikulum, stabilnost i homogenost.
3) Eksperimentalnim životinjama:
- korišćena vrsta/soj;
- broj, starost i pol životinja;
- izvor životinja, smeštaj, ishrana i itd.;
- Individualne telesne mase životinja na početku testa.
4) Uslovima ispitivanja:
- detalji primene ispitivanog preparata (mesto primene, analitičke metode, okluzija/neokluzija, zapremina, ekstrakcija, detekcija);
- detalji o kvalitetu hrane i vode.
5) Rezultatima:
- svi znaci toksičnosti;
- tabelarni podaci o apsorpciji (izraženi kao brzina, količina ili procenat);
- ukupni recovery u eksperimentu;
- tumačenje rezultata, poređenje sa raspoloživim rezultatima perkutane apsorpcije ispitivanog jedinjenja.
6) Obrazloženju rezultata.
7) Zaključcima.
1. Testing Method B.45. Skin Absorption: In vitro Method.
2. OECD (2002). Guidance Document for the Conduct of Skin Absorption Studies. OECD, Paris.
3. ECETOC, (1993) Percutaneous Absorption. European Centre for Ecotoxicology and Toxicology of Chemicals, Monograph No 20.
4. Zendzian R.P. (1989) Skin Penetration Method suggested for Environmental Protection Agency Requirements. J. Am. Coll. Toxicol. 8(5), p. 829-835.
5. Kemppainen, B.W., Reifenrath WG (1990) Methods for skin absorption. CRC Press Boca Raton, FL, USA.
6. EPA, (1992) Dermal Exposure Assessment: Principles and Applications. Exposure Assessment Group, Office of Health and Environmental Assessment.
7. EPA, (1998) Health Effects Test Guidelines, OPPTS 870-7600, Dermal Penetration. Office of Prevention, Pedsticides and Toxic Substances.
8. Bronaugh, R.L., Wester, R.C., Bucks, D., Maibach H.I. and Sarason, R., (1990) In vivo percutaneous absorption of fragrance ingredients in reshus monkeys and humans. Fd. Chem. Toxic. 28, p. 369-373.
9. Feldman, R.J. and Maibach, H.I., (1970) Absorption of some organic compounds through the skin in man. J. Invest Dermatol. 54, p. 399-404.
Slika 1: Primer dizajna tipične naprave koja se koristi da odredi i zaštiti mesto dermalne primene za vreme in vivo studija perkutane apsorpcije.
B.45. APSORPCIJA PREKO KOŽE: IN VITRO METODA
Ova metoda potpuno odgovara metodi OECD: TG 428 (2004).
Ova metoda je namenjena da obezbedi podatke o apsorpciji ispitivane supstance koja je primenjena na odsečak kože. Ona može da se kombinuje sa in vivo metodom za apsorpciju kroz kožu1 ili da se izvodi odvojeno. Preporučuje se da se koristi OECD vodič za izvođenje ispitivanja apsorpcije kroz kožu2 kao pomoć u dizajniranju studije zasnovane na ovoj metodi. Vodič je pripremljen da olakša izbor odgovarajućih in vitro postupaka za korišćenje u specifičnim okolnostima, da bi se obezbedila pouzdanost rezultata dobijenih ovom metodom.
Metode za merenje apsorpcije kroz kožu i dermalnog transporta mogu da se podele u dve kategorije: in vivo i in vitro. In vivo metode mogu da obezbede dobar farmakokinetički podatak o apsorpciji kroz kožu kod različitih vrsta životinja. In vivo metoda je opisana u posebnoj metodi1. In vitro metode se godinama koriste za merenje apsorpcije kroz kožu. Mada formalne validacione studije in vitro metoda koje se nalaze u ovoj metodi ispitivanja nisu izvedene, OECD eksperti su se složili 1999. godine da postoji dovoljno evaluiranih podataka koji opravdavaju in vitro metodu3. Dalji podaci koji potvrđuju ovo, uključujući i značajan broj poređenja in vivo i in vitro metoda navedeni su u vodiču2. Postoje brojne monografije koje razmatraju ovu temu i obezbeđuju detaljnu pozadinu primene in vitro metode4, 5, 6, 7, 8, 9, 10, 11, 12. In vitro metode mere difuziju hemikalija u i kroz kožu do rezervoara tečnosti i mogu da koriste neživu kožu za merenje difuzije ili svežu, metabolički aktivnu kožu za istovremeno merenje difuzije i metabolizma kože. Ove metode su posebno pogodne kao skrining za poređenje transporta hemikalija u i kroz kožu iz različitih formulacija i mogu da obezbede korisne modele za procenu perkutane apsorpcije kod ljudi.
In vitro metoda nije primenjiva u svim situacijama i za sve klase hemikalija. Moguće je koristiti in vitro metodu ispitivanja za inicijalnu kvalitativnu evaluaciju penetracije kroz kožu. U određenim slučajevima može biti neophodno da se ovo isprati podacima dobijenim u in vivo ispitivanjima. Vodič2 treba konsultovati za razjašnjenje situacija u kojima bi in vitro metoda bila pogodna. Dodatni detaljni podaci obezbeđeni su u dokumentu3.
Ova metoda prikazuje opšte principe za merenje dermalne apsorpcije i transporta ispitivane supstance uz upotrebu isečka kože. Može da se koristi koža mnogih vrsta sisara uključujući i ljudsku. Karakteristika permeabilnosti kože se zadržava i posle isecanja zato što je glavna difuziona barijera mrtvi stratum corneum, a aktivni transport hemikalija kroz kožu nije do sada identifikovan. Za kožu je pokazano da može da metaboliše neke hemikalije tokom perkutane apsorpcije6, ali ovaj proces nije ograničavajući u smislu apsorbovane doze, mada može da utiče na prirodu materijala koji ulazi u cirkulaciju.
Neapsorbovana doza jeste doza koja je isprana sa površine kože posle ekspozicije i deo koji je prisutan na neokluzivnom pokrivaču, uključujući i dozu za koju je pokazano da je isparila sa kože tokom ekspozicije.
Apsorbovana doza (in vivo) jeste količina ispitivane supstance koja je dospela u prijemnu tečnost ili sistemsku cirkulaciju tokom navedenog vremenskog perioda.
Apsorbabilna doza jeste doza na ili u koži posle ispiranja.
Ispitivana supstanca, najbolje radioaktivno obeležena, primenjuje se na površinu uzorka kože koja razdvaja dve komore difuzione ćelije. Hemikalija ostaje na koži određeno vreme sa kožom pod određenim uslovima, pre uklanjanja odgovarajućim postupkom čišćenja. Uzorci prijemne tečnosti se uzimaju u određenim vremenskim instancama tokom eksperimenta i analiziraju se na ispitivanu hemikaliju i/ili metabolite.
Kada se koriste metabolički aktivni sistemi, mogu da se analiziraju metaboliti ispitivane hemikalije pomoću odgovarajućih metoda. Na kraju eksperimenta kvantifikuje se distribucija ispitivane hemikalije i njenih metabolita kada to odgovara.
Koristeći odgovarajuće uslove, koji su opisani u ovoj metodi i u vodiču2, apsorpcija ispitivane supstance tokom datog perioda se meri analizom prijemne tečnosti i tretirane kože. Ispitivana supstanca koja ostaje u koži treba da se smatra apsorbovanom, osim ako može da se pokaže da apsorpcija može da se odredi samo iz vrednosti u prijemnoj tečnosti. Analiza drugih komponenti (materijal ispran sa kože i onaj koji je ostao u slojevima kože) omogućuje dalju evaluaciju podataka, uključujući ukupnu dispoziciju supstance i recovery u procentima.
Da bi pokazali mogućnosti i pouzdanost test sistema u laboratoriji u kojoj se izvodi ispitivanje, rezultati za relevantne referentne hemikalije treba da budu dostupni i da budu u skladu sa podacima datim u literaturi za metodu koja se koristi. Ovaj zahtev može da se ispuni ispitivanjem odgovarajuće referentne supstance (poželjno je da lipofilnost bude bliska lipofilnosti ispitivane supstance), istovremeno sa ispitivanom supstancom ili obezbeđivanjem odgovarajućih istorijskih podataka o većem broju referentnih supstanci različite lipofilnosti (nrp. kofein, benzoeva kiselina i testosteron).
1.4.1. Difuziona ćelija
Difuziona ćelija se sastoji od donorske komore i prijemne komore između kojih se postavlja koža (primer tipičnog dizajna je dat na Slici 1.). Ćelije treba da obezbede dobro zaptivanje oko kože, omoguće jednostavno uzorkovanje i dobro mešanje prijemnog rastvora u kontaktu sa donjom stranom kože kao i dobru kontrolu temperature ćelije i njenog sadržaja. Statične i protočne difuzione ćelije su jednako prihvatljive.
Obično se donorske ćelije ostavljaju neokludirana za vreme ekspozicije određenoj dozi ispitivanog preparata. Za neodređene aplikacije i pojedina scenarija za određene doze, donorske komore mogu da se okludiraju.
1.4.2. Prijemna tečnost
Preporučuje se korišćenje fiziološki pogodne prijemne tečnosti, mada mogu da se koriste i druge pod uslovom da je to opravdano. Navodi se precizni sastav prijemne tečnosti. Odgovarajuća rastvorljivost ispitivane hemikalije u prijemnoj tečnosti treba da se pokaže tako da tečnost ne predstavlja barijeru za apsorpciju. Uz to, prijemna tečnost ne treba da utiče na integritet preparata kože. U protočnom sistemu brzina protoka ne sme da utiče na difuziju ispitivane supstance u prijemnu tečnost. U statičnom ćelijskom sistemu, prijemna tečnost treba da se neprekidno meša i redovno uzorkuje. Ako se ispituje metabolizam, prijemna tečnost treba da obezbeđuje da koža ostane živa tokom celokupnog eksperimenta.
1.4.3. Preparati kože
Može da se koristi koža od ljudi ili životinja. Poznato je da korišćenje ljudske kože je predmet nacionalnih i internacionalnih etičkih razmatranja i uslova. Mada je živa koža bolja, neživa koža može da se koristi pod uslovom da može da se pokaže integritet kože. Prihvatljive su epidermalne membrane (enzimski, toplotno ili hemijski odvojene), ili odvojeni slojevi kože (obično debljine 200-400 µm) pripremljeni sa dermatomom. Puna debljina kože može da se koristi ali prekomerna debljina (približno više od 1 mm) treba da se izbegava sem u slučaju specifičnog zahteva da se odredi ispitivana hemikalija u slojevima kože. Izbor vrste, anatomskog mesta i tehnika pripreme mora biti opravdana. Potrebni su prihvatljivi podaci od najmanje četiri ponavljanja po ispitivanom preparatu.
1.4.4. Integritet preparata kože
Suštinsko je da se koža dobro pripremi. Neodgovarajuće rukovanje može da ima za posledicu oštećenje stratus corneuma i zbog toga integritet pripremljene kože mora da se proveri. Kada se ispituje metabolizam kože, svež isečak kože treba da se koristi što je pre moguće u uslovima za koje se zna da održavaju metabolizam kože. Kao opšte pravilo, svež isečak kože treba da se koristi u okviru 24 sata, ali prihvatljiv period čuvanja može da varira zavisno od enzimskog sistema koji je uključen u metabolizam i temperature čuvanja13. Kada se preparati kože čuvaju pre upotrebe, mora da postoji dokaz da je funkcija barijere očuvana.
1.4.5. Ispitivana supstanca
Ispitivana supstanca je entitet čiji penetracione karakteristike treba da se ispitaju. Idealno, ispitivana supstanca treba da bude radioaktivno obeležena.
1.4.6. Ispitivani preparat
Preparat ispitivane supstance (npr. čista, razblažena ili formulisani materijal koji sadrži ispitivanu hemikaliju koja se primenjuje na kožu), treba da bude jednak (ili realna zamena), kao onaj kojem će biti eksponirani ljudi ili druga moguća ciljna vrsta. Svaka varijacija od preparata u upotrebi mora biti opravdana.
1.4.7. Koncentracije ispitivane supstance i formulacije
Obično više od jedne koncentracije ispitivane supstance koristi se odražavajući gornju ili potencijalnu ekspoziciju ljudi. Treba razmotriti ispitivanje različitih tipičnih formulacija.
1.4.8. Primena na kožu
U uobičajenim uslovima ljudske ekspozicije hemikalijama sreću se određene doze. Količina koja se primenjuje treba da imitira moguću ekspoziciju kod ljudi, tipično 1 mg/cm2 do 5 mg/cm2 kože za čvrste ili 10 µl/cm2 za tečnosti.
Količine treba da se opravdaju očekivanim uslovima primene, ciljevima studije ili fizičkim svojstvima ispitivanog preparata. Na primer, primene na površinu kože mogu biti neodređene u slučajevima gde se nanose velike zapremine po jedinici površine.
1.4.9. Temperatura
Na pasivnu difuziju hemikalija (i apsorpciju kroz kožu) utiče temperatura. Difuzionu komoru i kožu treba održavati na konstantnoj temperaturi koja je bliska temperaturi kože od 32 °C ± 1 °C. Različiti dizajn ćelija ima različite uslove vodenog kupatila ili temperaturnog bloka za obezbeđivanje održavanja temperature prijemnika i kože u fiziološkim granicama. Vlažnost treba da bude između 30% i 70%.
1.4.10. Trajanje ekspozicije i uzorkovanje
Ekspozicija kože ispitivanom preparatu može biti tokom celokupnog eksperimenta ili kraćeg perioda (tj. da bi se imitirala specifična vrsta ekspozicije ljudi). Koža treba da bude isprana od viška ispitivanog preparata odgovarajućim sredstvom za čišćenje, a ispirak skupljen za analizu. Postupak uklanjanja ispitivanog preparata zavisi od očekivanog uslova kod primene i treba da bude opravdana. Period uzorkovanja od 24 sata je uobičajen da bi se obezbedila adekvatna karakterizacija profila apsorpcije. S obzirom da se integritet kože posle 24 sata narušava, vremena uzorkovanja ne treba da budu duža od 24 sata. Za ispitivane supstance koje prodiru u kožu brzo, ovo možda nije neophodno, ali za supstance koje prodiru sporo može da bude potrebno duže vreme. Učestalost uzimanja uzoraka prijemne tečnosti obezbeđuje grafičko prikazivanje apsorpcionog profila ispitivane supstance.
1.4.11. Završni postupci
Svi delovi ispitivanog sistema treba da se analiziraju i da se odredi recovery. Ovo uključuje donorsku komoru, ispirak površine kože, preparat kože i prijemnu tečnost/komoru. U nekim slučajevima koža može da se razdvoji na izloženi deo i deo kože ispod ćelijskog oboda i na stratum corneum, epidermis i dermis radi zasebnih analiza.
1.4.12. Analiza
U svim studijama treba postići odgovarajući recovery (npr. 100% ± 10% radioaktivnosti i svako odstupanje treba da bude opravdano). Količina ispitivane supstance u prijemnoj tečnosti, preparatu kože, ispicima površine kože i ispirak aparature treba da se analizira korišćenjem odgovarajuće tehnike.
Navode se: analiza prijemne tečnosti, distribucija ispitivane hemikalije u test sistemu i apsorpcioni profil u vremenu. Kada se koriste uslovi određene doze, treba da se izračuna: količina isprana sa kože, količina združena sa kožom (i u različitim slojevima kože, ako se analizira) i količina prisutna u prijemnoj tečnosti (brzina, i količina ili procenat od primenjene doze). Apsorpcija kroz kožu može ponekad da se izrazi korišćenjem podataka dobijenih samo analizom prijemne tečnosti. Kada ispitivana supstanca ostane u koži na kraju studije, može da bude potrebno da se ta količina uključi u ukupnu apsorbovanu količinu (videti literaturu3). Kada se u ekspoziciji koriste uslovi neodređene doze, podaci će omogućiti izračunavanje konstante permeabilnosti (u daljem tekstu: Kp). Pod ovim uslovima, procenat koji je apsorbovan nije relevantan.
Izveštaj o ispitivanju sadrži zahteve navedene u protokolu uključujući opravdanost sistema za testiranje koji je korišćen, a sadrži i podatke o:
1) Ispitivanoj supstanci:
- fizička svojstva, fizička i hemijska svojstva (najmanje molekulska masa i log Pow), čistoća (radiohemijska čistoća);
- podaci o identifikaciji (npr. broj serije);
- rastvorljivost u prijemnoj tečnosti.
2) Ispitivanom preparatu:
- formulacija i opravdanost primene;
- homogenost.
3) Uslovima ispitivanja:
- izvori i predeo kože, metod pripreme, uslovi čuvanja pre upotrebe, pretretman (čišćenje, tretman antibioticima, itd.), merenja integriteta kože, metabolički status, opravdanost upotrebe;
- dizajn ćelije, sastav prijemne tečnosti, brzina protoka prijemne tečnosti ili vremena uzorkovanja i postupci;
- detalji primene ispitivanog preparata i kvantifikacija primenjene doze;
- trajanje ekspozicije;
- detalji uklanjanja ispitivanog preparata sa kože, tj. ispiranje kože;
- detalji analize kože i tehnika frakcionisanja koje su korišćene da pokažu distribuciju u koži;
- postupci za pranje ćelija i opreme;
- metode za analizu, tehnike ekstrakcije, limiti detekcije i validacija analitičke metode.
4) Rezultatima:
- ukupan recovery eksperimenta (primenjena doza = ispirci kože + koža + prijemna tečnost + ispirci ćelija);
- tabeliranje individualnih recovery vrednosti ćelija za svaki kompartman;
- apsorpcioni profil;
- tabelirani podaci o apsorpciji (izraženi kao brzina, količina ili procenat).
5) Obrazloženju rezultata.
6) Zaključcima.
1. Testing Method B.44. Skin Absorption: In vivo Method.
2. OECD, (2002) Guidance Document for the Conduct of Skin Absorption Studies. OECD, Paris.
3. OECD, (2000) Report of the Meeting of the OECD Extended Steering Committee for Percutaneous Absorption Testing, Annex 1 to ENV/JM/TG(2000)5. OECD, Paris.
4. Kemppainen B.W. and Reifenrath W.G., (1990) Methods for skin absorption. CRC Press, Boca Raton.
5. Bronaugh R.L. and Collier, S.W., (1991) Protocol for In vitro Percutaneous Absorption Studies, in In vitro Percutaneous Absorption: Principles, Fundamentals and Applications, RL Bronaugh and HI Maibach, Eds., CRC Press, Boca Raton, p. 237-241.
6. Bronaugh R.L. and Maibach H.I., (1991) In vitro Percutaneous Absorption: Principles, Fundamentals and Applications. CRC Press, Boca Raton.
7. European Centre for Ecotoxicology and Toxicology of Chemicals, (1993) Monograph No 20, Percutaneous Absorption, ECETOC, Brussels.
8. Diembeck W, Beck H, Benech-Kieffer F, Courtellemont P, Dupuis J, Lovell W, Paye M, Spengler J, Steiling W., (1999) Test Guidelines for In vitro Assessment of Dermal Absorption and Percutaneous Penetration of Cosmetic Ingredients, Fd Chem Tox, 37, p. 191-205.
9. Recommended Protocol for In vitro Percutaneous Absorption Rate Studies (1996). US Federal Register, Vol. 61, No 65.
10. Howes, D., Guy, R., Hadgraft, J., Heylings, J.R. et al. (1996). Methods for assessing Percutaneous absorption. ECVAM Workshop Report ATLA 24, 81 R10.
11. Schaefer, H. and Redelmeier, T.E., (1996). Skin barrier: principles of percutaneous absorption. Karger, Basel. (12) Roberts, M.S. and Walters, K.A., (1998). Dermal absorption and toxicity assessment. Marcel Dekker, New York.
13. Jewell, C., Heylings, J.R., Clowes, H.M. and Williams, F.M. (2000). Percutaneous absorption and metabolism of dinitrochlorobenzene in vitro. Arch Toxicol 74, p. 356-365.
Slika 1: Primer tipičnog dizajna stičke difuzione ćelije za in vitro studije perkutane apsorpcije.